Alternative synthesis of the anti-baldness compound RU58841†
Matthew J.
Leonard
,
Anthony R.
Lingham
,
Julie O.
Niere
,
Neale R. C.
Jackson
,
Peter G.
McKay
and
Helmut M.
Hügel
RMIT University, 1 Bowen street, Melbourne 3001, Australia. E-mail: Matthew.Leonard742@gmail.com
First published on 6th March 2014
Abstract
RU58841 is active against baldness and is commercially available. The previously reported synthesis uses phosgene, three discrete inert atmosphere steps and three steps that require flash chromatography. Our synthesis uses no phosgene, only one inert atmosphere step and does not require flash chromatography. This is achieved by stepwise construction of the hydantoin moiety around the amino group of 3-trifluoromethyl-4-cyanoaniline and ring closure to give a 2-nitropropane leaving group. On a small scale we achieved an overall yield of 33%.
Introduction
Hydantoins are five-membered heterocycles with four points of functionality (Fig. 1). | ||
| Fig. 1 Hydantoin group and RU58841 anti-baldness compound. | ||
They are a staple scaffold displayed in many compounds with functional end use such as cosmetics, shampoos and skin lotions. Important medicinal compounds that incorporate the hydantoin moiety include phenytoin (antiepileptic) and nilutamide (anticancer)1 (Fig. 2). The chiral hydantoin compound (+)-hydantocidin (Fig. 2) is extracted from Streptomyces hygroscopicus and is a potent herbicide.2
 | ||
| Fig. 2 Biologically-active hydantoin compounds. | ||
Given the common occurrence of hydantoins in a diverse range of compounds, alternative hydantoin syntheses are potentially useful. Recent additions include access to 5,5-disubstituted hydantoins by treatment of nitriles with an organometallic reagent (RLi or RMgX) followed by KCN/(NH4)2CO3 (ref. 1) and use of carbodiimides to prepare intermediates that form hydantoins by ring closure and rearrangement.3
In the 1970s, T. Battmann and co-workers at the French pharmaceutical company Roussel-Uclaf explored hydantoin compounds to treat a number of ailments. They prepared RU58841 (Fig. 1), which was targeted as a prostate cancer treatment,4 but biological testing with rats produced hair growth. Roussel-Uclaf prepared homologues of RU58841 (Fig. 3) including RU58642 (a hydantoin) and RU56187 (a thiohydantoin) which were both found to be active non-steroidal anti-androgens.5
 | ||
| Fig. 3 RU58642 and RU56187. | ||
While RU58642 had a stronger anti-androgen effect than RU58841, its side effects made it unusable in patients. RU58642 has since found use in biochemical research into the androgen receptor.6,7
Research interest in anti-androgen pharmaceuticals thrived in the following decades. In 1994 Battmann et al. published biological evaluation of both RU58841 and RU56187 (ref. 5) and their synthesis of RU58841.4 They prepared the hydantoin by first reacting 3-trifluoromethyl-4-cyanoaniline with phosgene to produce an isocyanate intermediate (Fig. 4). The 1994 paper4 reported that the dose required for hair regrowth was only one third of that required to give systemic side effects making RU58841 viable as a topical anti-androgen.4 Further work by De Brouweri et al. confirmed the effect of RU58841 as an anti-androgen and baldness treatment by observing hair re-growth on bald human scalp segments that had been grafted to mice.8 As well as empirical studies, the biochemistry of RU58841 and other hydantoins have been evaluated for their effect as non-steroidal testosterone inhibitors.9
 | ||
| Fig. 4 Traditional synthesis of RU58841. | ||
As the synthesis of RU58841 by Battmann et al. (Fig. 4) uses phosgene (a toxic gas which requires a high level of expertise with handling), alternative preparations have been sought. In 2006 Hügel et al. prepared RU58841 by N-arylation of 5,5-dimethylhydantoin.10 They achieved the RU58841 hydantoin synthon in 55% yield by NaH/halogen aryl-coupling of 5,5-dimethylhydantoin with 3-trifluoromethyl-4-cyanoaniline but the yield lowered to 30% when carried out on a 10 g scale. Replacing this NaH coupling with copper acetate promoted boronic acid coupling gave a 79% yield of the same synthon.10 When this method was tried using other anilines the yields were limited to ∼50% and yields above 60% could only be achieved when the aniline being attached to the hydantoin had either nitrile or methoxy substituents.11
To improve on the RU58841 syntheses by Battmann et al. and Hugel et al., in lieu of aryl-coupling we now present an approach which uses the same start material as Battmann et al.4 (3-trifluoromethyl-4-cyanoaniline) but builds the hydantoin moiety around the aniline so as to avoid the carbonylation step which requires either the highly toxic gas phosgene or phosgene equivalents reagents which are difficult to upscale and still somewhat toxic. This provides a more flexible synthesis than the boronic acid coupling approach by Hugel et al. as it is transferable to the preparation of a wider variety of other hydantoin target compounds.
Results and discussion
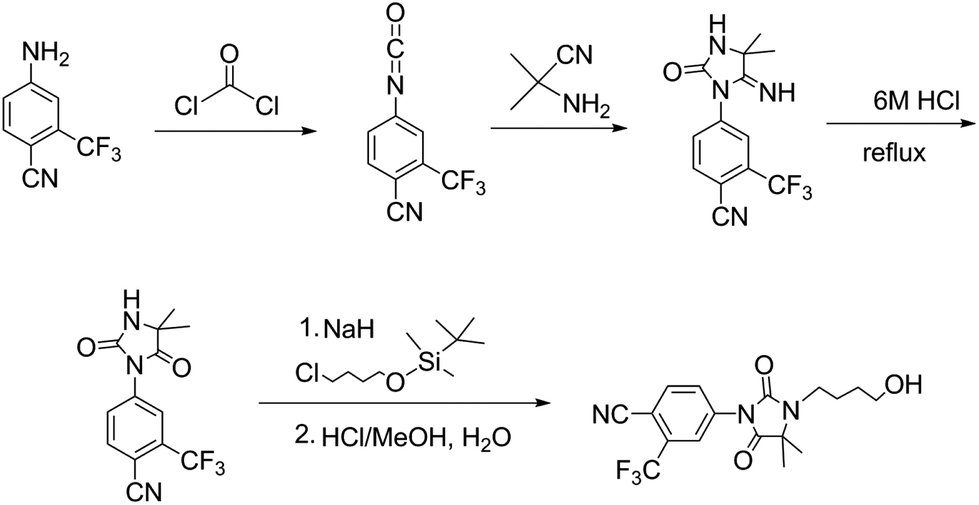
Starting from 3-trifluoromethyl-4-cyanoaniline, our six step synthesis prepares a long chain double-amide intermediate which undergoes a ring closure to give the stable five membered hydantoin moiety (compound 6) which yields 2-nitropropane as a side product. The hydantoin 6 is then alkylated with 4-bromobutyl acetate using NaH/halogen coupling and deprotected by addition of NaOH to the same pot to furnish the product – RU58841 (Fig. 5). | ||
| Fig. 5 Alternative synthesis of RU58841. | ||
During our efforts to access the RU58841 synthon 6 without using phosgene we discovered that, unexpectedly, reaction of the tertiary bromide 1 with excess sodium nitrite in DMF at room temperature produced an 86% yield of the stable nitro compound 2. Such a reaction at a tertiary carbon appears to have no literature precedent. The generality of this synthetically useful reaction is being explored and will be reported soon.
Upon discovery that the bromine had been substituted with a nitro group, this appeared a safer and more convenient approach to access to the desired hydantoin 6 for RU58841 than the synthesis reported by Battmann et al. (Fig. 4). In keeping with our aim to use less toxic reagents that are easy to handle, this new scheme (Fig. 5) achieves the hydantoin 6 and thence RU58841 by way of ‘reacylation’ of 3, bromo-nitro substitution to furnish 5in situ and then a ring closure of 5 with 2-nitropropane acting as the leaving group. Thus we present a simple new hydantoin preparation exemplified by the synthesis of compound RU58841 in 33% overall yield.
Bromine-nitro substitution
Compound 2 was prepared by placing 1 in DMF with a 4–10 molar excess of NaNO2. It was stirred at RT for 14 h with no effort made to keep anhydrous conditions. When the reaction was seen to be complete by TLC, a volume of water equal to the volume of DMF used was added and the flask allowed to sit for 1 hour. For a more complete crystallization, the flask was placed into a fridge at 8 °C overnight. The crystals were filtered for an apparent 122% yield which was due to the co-crystallization of DMF with the product in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.†
:1 ratio.†
Compound 2 is an α-nitroisobutyranilide. This class of compounds has been described before only once12 where they were prepared by a different and more complex route. Bromine-nitro substitution from exposure to nitrite ions is unexpected for compound 1, as SN2 reactions at tertiary carbons are virtually unknown,13 while SN1 reactions of nitrite normally give nitrite esters rather than nitro compounds.13 We believe neither mechanism to be operative here. This general reaction has been explored and will be discussed elsewhere.
Ring closure to prepare hydantoin compound (6)
A number of room temperature methods with catalysts were tried, however the cleanest method for ring closure of 5 proved to be heating while still in the DMF from the previous step at 110 °C for 7 h. GC-MS showed a major peak (89% of total peak area). Workup gave a white solid (compound 6, 92% crude yield) that was used for further reaction without recrystallization.This ring closure provides a new way to prepare N-aryl hydantoins. It also yields 2-nitropropane as a leaving group which is of interest as it has not (to the best of our knowledge) been reported as a leaving group before.
Two alternative mechanisms can be envisaged for this cyclisation both of which adhere to the Baldwin rules. The anilide nitrogen could perform an acyl substitution at the other carbonyl in a 5-exo-trig attack, with loss of 2-nitropropane anion. However the high steric hindrance about this amide carbonyl makes this seem unlikely. Alternatively, the 2-nitropropane anion could be lost first to create a much more electrophilic, much less hindered isocyanate, which would then be attacked in a 5-exo-dig geometry (Fig. 6).
 | ||
| Fig. 6 Probable mechanism for hydantoin formation. | ||
This mechanism is supported by our observation that the α-nitroisobutyranilide 2 appears to be converted by injection into a conventional mass spectrometer into the corresponding isocyanate, with loss of 2-nitropropane. The dipolar/partial ionic nature of DMF may help to stabilize the amide and encourage the anilide nitrogen to proceed via the isocyanate mechanistic pathway. This represents a novel isocyanate preparation and will be further studied to be reported elsewhere.
Comparison of RU58841 and its synthon (6) with literature data
The RU58841 and compound 6 prepared by us had identical UV and IR properties to those reported by Battmann et al.4 Our 1H NMR chemical shifts for each compound differed slightly from those reported; this may be due to a difference in solvent as Battmann et al. did not report their solvent or compound concentration. We report the 13C NMR data for both compounds for the first time. Our melting point for 6 matched that reported, but we have repeatedly obtained a melting point for RU58841 of 71–72 °C from highly crystalline samples that are pure by C, H, N analysis, while Battmann et al. report 103–104 °C. Our 1H and 13C NMR of RU58841 gave three minor peaks which could not be assigned to the compound at δ 6.7 in 1H NMR and at δ 116.3 and δ 151 in the 13C NMR. These are possible due to a tautomeric impurity present at very low levels, such that it does not affect the results of C, H, N analysis.Conclusions
We have added to the range of hydantoin syntheses with a synthesis of RU58841 in a 33% overall yield. An α-isobutyryl bromide exposed to nitrite ions was shown to readily and cleanly convert to its equivalent α-isobutyryl nitro compound. This demonstrates that bromine-nitro substitutions do occur on tertiary carbons with a neighbouring carbonyl. 2-Nitropropane was shown to be an effective leaving group, enabling the formation of a hydantoin ring by cyclization. This method of achieving a hydantoin ring may be used to prepare a suite of hydantoins for testing or to achieve an alternative end-use hydantoin.Experimental section
General methods
Chemicals were used unaltered from the following suppliers: 3-trifluoromethyl-4-cyanoaniline (AK Scientific); α-bromoisobutyryl bromide (Sigma-Aldrich); 1,2-dichloroethane (BDH); toluene–n-heptane–NaNO2 (Ajax); DMF–ethyl acetate–methanol (Chem-Supply); tert-butyl methyl ether–pentane (Merck). K2CO3 (Ajax) was stored in oven at 115 °C before use. TLC plates from Merck were labelled “TLC silica gel 60 F254”. The dry DMF used for step 7 was obtained from a solvent dispensing system immediately before use, but DMF used in steps 2 and 5 was used unaltered from the bottle (Chem-Supply).IR spectra were measured on a Varian 1000 FTIR spectrometer as KBr disks (4000–400 cm−1) with images provided in the ESI.†1H and 13C NMR spectra were recorded on a Bruker 300 MHz spectrometer (images provided in the ESI†). Chemical shifts in 1H NMR spectra were reported in parts per million (ppm δ) relative to the TMS signal and were measured using the residual chloroform solvent signal set to 7.24 ppm. Chemical shifts in 13C NMR spectra were measured relative to the central peak of the deuterochloroform signal (δ = 77.5 ppm). Coupling constants were reported in Hz. Low-resolution mass spectra for monitoring and compound confirmation were carried out on a Varian CP-3800 GC connected to a Varian Saturn 2200 GC-MS-MS equipped with a 30 m SGE BPX5 Column and also on a Micromass Platform II electrospray using MassLynx software. High-resolution mass spectra for compounds 1–5 were carried out at Monash University on an Agilent 6220 accurate mass LC-TOF system equipped with an Agilent 1200 series HPLC column. High-resolution mass spectra for compound 6 and RU58841 were carried out at RMIT on a Waters GCT Premier HR-TOFMS equipped with an Agilent 7890 GC column. Crystallography was carried out on a Bruker APEX II DUO diffractometer.
2-Bromo-N-[4-cyano-3-(trifluoromethyl)phenyl]-2-methylpropanamide (compound 1)
3-Trifluoromethyl-4-cyanoaniline (3.00 g, 16.1 mmol) was dissolved in 1,2-dichloroethane (35 mL) and placed in a flask that contained oven-dried K2CO3 (2.00 g), to keep the mixture anhydrous and basic. α-Bromoisobutyryl bromide (4.10 g, 17.8 mmol) was added and the mixture stirred at RT for 14 h. 1,2-Dichloroethane was removed and solids worked up in EtOAc–water to yield 5.30 g (99%) of 1, which recrystallized from hot methanol to give 4.85 g (14.5 mmol) of clear light brown cubic crystals, mp 127–129 °C; yield 90%; Rf = 0.50 in 4: 1 hexanes–EtOAc; IR (cm−1): 3293, 3099, 3057, 2987, 2935, 2234 (CN), 1666 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1586, 1520, 1427, 1325, 1185 & 1139 (C–F), 1052, 847, 672; 1H NMR (300 MHz, CDCl3): δ 2.06 (6H, s, CH3), δ 7.82 (d, ArH5, J 8), δ 7.92 (dd, ArH6, J 2, J 8), δ 8.05 (d, ArH2, J 2), δ 8.73 (br, s, NH); 13C NMR (75 MHz, 40 mg: 0.4 mL DMSOd6): δ 31.3 (s, C-3A/B), δ 60.6 (s, C-2), δ 103.3 (s, C-4′), δ 116.7 (s, CN), δ 118.5 (q, C-2′, J 5), δ 123.4 (q, CF3, J 136), δ 123.7 (s, C-6′), δ 132.6 (q, C-3′, J 32), δ 137.1 (s, C-5′), δ 144.4 (s, C-1′), δ 171.3 (s, C-1); Neg ESI HRMS: calcd for C12H10N2OF3Br (M − H): 332.9850, observed: m/z 332.9858.†
O), 1586, 1520, 1427, 1325, 1185 & 1139 (C–F), 1052, 847, 672; 1H NMR (300 MHz, CDCl3): δ 2.06 (6H, s, CH3), δ 7.82 (d, ArH5, J 8), δ 7.92 (dd, ArH6, J 2, J 8), δ 8.05 (d, ArH2, J 2), δ 8.73 (br, s, NH); 13C NMR (75 MHz, 40 mg: 0.4 mL DMSOd6): δ 31.3 (s, C-3A/B), δ 60.6 (s, C-2), δ 103.3 (s, C-4′), δ 116.7 (s, CN), δ 118.5 (q, C-2′, J 5), δ 123.4 (q, CF3, J 136), δ 123.7 (s, C-6′), δ 132.6 (q, C-3′, J 32), δ 137.1 (s, C-5′), δ 144.4 (s, C-1′), δ 171.3 (s, C-1); Neg ESI HRMS: calcd for C12H10N2OF3Br (M − H): 332.9850, observed: m/z 332.9858.†
N-[4-Cyano-3-(trifluoromethyl)phenyl]-2-methyl-2-nitropropanamide (compound 2)
Compound 1 (5.00 g, 15.0 mmol) was dissolved in DMF (130 mL) and NaNO2 (10.0 g, 145 mmol) was added. The contents were stirred at room temperature for 14 h then transferred to a larger flask where deionized water (130 mL) was added which gave heat and produced white needles that were filtered and washed with water to give 4.84 g of 2 that were shown to be co-crystallized in a 1:1 ratio with DMF and therefore contained 12.9 mmol of 2 for a corrected yield of 86%, mp 129–131 °C (co-crystallized with DMF); Rf = 0.79 in 1:1 hexanes–EtOAc or 0.17 in 4:1 hexanes–EtOAc; DMF removed by repeated water–EtOAc extraction for IR (cm−1): 3337, 3191, 3119, 3062, 3003, 2948, 2237 (CN), 1706 (CO), 1603, 1562, 1536, 1504, 1323, 1180 & 1134 (C–F), 1050, 895, 857, 640, 558; 1H NMR (300 MHz, 20 mg: 0.4 mL CDCl3): δ 2.02 (6H, s, CH3), δ 3.53 (br, s, NH), δ 8.18 (d, ArH5, J 8), δ 8.26 (dd, ArH6, J 2, J 8), δ 8.40 (d, ArH2, J 2), δ 10.80 (s, NH); 13C NMR (75 MHz, 40 mg: 0.4 mL CDCl3): δ 23.7 (s, C-3A/B), δ 92.1 (s, C-2), δ 103.8 (s, C-4′), δ 115.9 (s, CN), δ 118.1 (q, C-2′, J 5), δ 123.0 (q, CF3, J 136), δ 123.5 (s, C-6′), δ 132.5 (q, C-3′, J 17), δ 136.7 (s, C-5′), δ 143.5 (s, C-1′), δ 167.4 (s, C-1); Neg ESI HRMS: calcd for C12H10N3O3F3 (M − H): 300.0596, observed: m/z 300.0602.†
2-Amino-N-[4-cyano-3-(trifluoromethyl)phenyl]-2-methylpropanamide (compound 3)
Compound 2 (5.00 g, 13.4 mmol) was placed in a flask and solvents were added, namely ethanol (32 mL), water (18 mL) and 2-propanol (9 mL). Fe0 powder (1.50 g, 35.8 mmol) was added and the reaction taken to reflux (90 °C) at which time ∼1 mL of [32%] hydrochloric acid was added through the top of the condenser. After 1 h the reaction was allowed to cool and TLC showed start material gone, replaced by a tailing spot typical of an amine.For the workup it should be noted that 3 is highly soluble in EtOAc. Another type of hydrogenation may be preferred to perform this reaction on large scale. Our workup was as follows: reaction mixture was vacuum filtered through celite to remove iron solids and the celite flushed with hot ethanol. Water–ethanol–2-propanol was removed by rotary evaporator and the solids dissolved in ethyl acetate–water. At pH < 7, compound 3 resides in the aqueous layer. Thus, at the initial pH ∼2, the ethyl acetate fraction was discarded, removing orange impurities. Conc. NaOH was added to the aqueous portion and a dark green/black Fe0 precipitate appeared at pH ∼5. The precipitate was removed by passing the mixture through celite. On addition of more NaOH, a white solid (3) precipitated. Ethyl acetate was added to dissolve this and the pH of the aqueous layer was brought to ∼10. When a little more Fe0 precipitate drifted to the bottom of the aqueous layer, this fraction and the solid were drawn off and discarded. The ethyl acetate fraction was then washed with another volume of deionised water before being dried with MgSO4 and again filtered through celite. The celite was rinsed with hot ethyl acetate. The solvent was removed from the combined ethyl acetate fractions by rotary evaporator to give 2.72 g (10.0 mmol) of 3 as clean white fluffy crystals, mp 113–115 °C; yield 75%; Rf = 0.11 in 1:1 hexanes–EtOAc; IR (cm−1): 3388, 3362, 3277 & 3233 (N–H), 2993, 2972, 2934, 2231 (CN), 1699 (CO), 1613, 1514, 1490, 1422, 1326, 1178 & 1138 (C–F), 1050, 909, 888, 858, 749, 734, 673, 558; 1H NMR (300 MHz, 75 mg: 0.5 mL DMSOd6): δ 1.34 (6H, s, CH3), δ 3.53 (br, s, NH), δ 5.05 (br, s, NH), δ 8.04 (d, ArH5, J 8), δ 8.18 (dd, ArH6, J 2, J 8), δ 8.43 (d, ArH2, J 2); 13C NMR (75 MHz, 75 mg: 0.4 mL DMSOd6): δ 29.2 (s, C-3A/B), δ 56.3 (s, C-2), δ 102.4 (s, C-4′), δ 116.8 (s, CN), δ 117.8 (q, C-2′, J 5), δ 123.1 (s, C-6′), δ 123.4 (q, CF3, J 136), δ 132.6 (q, C-3′, J 17), δ 137.2 (s, C-5′), δ 144.6 (s, C-1′), δ 179.1 (s, C-1); Neg ESI HRMS: calcd for C12H12N3OF3 (M − H): 270.0854, observed: m/z 270.0864.†
2-Bromo-N-(1-{[4-cyano-3-(trifluoromethyl)phenyl]carbamoyl}-1-methylethyl)-2-methylpropanamide (compound 4)
Compound 3 (1.03 g, 3.80 mmol) was dissolved in 1,2-dichloroethane (20 mL) in a flask that contained oven dried K2CO3 (1.00 g) and then stirred with α-bromoisobutyryl bromide (1.06 g, 4.06 mmol) for 14 h. 1,2-Dichloroethane was removed and the product worked up in EtOAc–water to yield 1.36 g (3.23 mmol) of 4 (85% yield) as an oil that solidified to an amorphous off-white foam after several hours at high vacuum.For crystallization, the solid was dissolved in dichloromethane and passed through a short column of silica gel, eluting with ethyl acetate. After evaporation of the solvent, the residue was further purified by extraction with boiling n-heptane. After cooling, the n-heptane was decanted off and the solid was recrystallized from m-xylene/n-pentane. Crystallization was slow and was completed overnight at 8 °C. The resulting crystals, (mp 121–123 °C) were pure enough for use in the next step, but contained co-crystallized m-xylene. To remove this, they were heated to 60 °C for 6 h under high vacuum, giving a white powder that could be converted to solvent-free crystals for crystallography (mp 107–109 °C) by very slow evaporation from toluene.
Mp 107–109 °C or 121–123 °C when co-crystallized with m-xylene; Rf = 0.78 in 1:1 hexanes–EtOAc or 0.07 in 4:1 hexanes–EtOAc; IR (cm−1): 3401, 3312 (N–H), 2992, 2932, 2229 (CN), 1722, 1664 (CO), 1611 (CO), 1512, 1427, 1328, 1174 & 1132 (C–F), 1049, 882, 850, 555; 1H NMR (300 MHz, 32 mg: 0.4 mL DMSOd6): δ 1.51 (6H, s, C-3/CH3), δ 1.94 (6H, s, C-6/CH3), δ 3.38 (NH-2), δ 8.09 (d, ArH5, J 8), δ 8.15 (dd, ArH6, J 2, J 8), δ 8.34 (d, ArH2, J 2), δ 10.03 (NH-1); 13C NMR (75 MHz, 32 mg: 0.4 mL DMSOd6): δ 24.8 (s, C-3A/B), δ 31.7 (s, C-5A/B), δ 58.2 (s, C-5), δ 61.7 (s, C-2), δ 102.3 (s, C-4′), δ 116.8 (s, CN), δ 117.8 (q, C-2′, J 5), δ 123.2 (s, C-6′), δ 123.5 (q, CF3, J 136), δ 132.4 (q, C-3′, J 17), δ 137.3 (s, C-5′), δ 146.0 (s, C-1′), δ 171.1 (s, C-4), δ 174.7 (s, C-1); Neg ESI HRMS: calcd for C16H17N3O2F3Br (M−): 420.0359, observed: m/z 420.0362.†
N-(1-{[4-Cyano-3-trifluoromethyl)phenyl]carbamoyl}-1-methylethyl-2-methyl-2-nitropropanamide (compound 5)
Compound 4 (1.00 g, 2.38 mmol) was dissolved in DMF (15 mL). NaNO2 (1.30 g, 18.8 mmol) added and the mixture stirred at RT for 14 h. The reaction was monitored by TLC and electrospray mass spectrometry. TLC showed that all compound 4 had reacted and a single different Rf of the newly formed compound. High resolution mass spectrometry confirmed the bromine had been replaced by a nitro group. The product was not isolated but used in situ for the next step.
R
f = 0.50 in 1:1 hexanes–EtOAc; neg ESI HRMS: calcd for C16H17N4O4F3Br (M − H): 385.1124, observed: m/z 385.1133.
4-(4,4-Dimethyl-2,5-dioxoimidazolidin-1-yl)-2-(trifluoromethyl)benzonitrile (compound 6)
The reaction vessel from the preparation of 5 was fitted with an air condenser and heated at 110 °C for 7 h,‡ the DMF was then removed by vacuum and the white solids worked up in water–ethyl acetate to obtain 587 mg (1.98 mmol) of 6 (83% yield). Longer reaction time or increased reaction temperature gave 6 in reduced yield that was harder to purify. For the purposes of obtaining mp, IR and NMR 6 was recrystallized from hot 2-propanol which lowered the yield to 40%.Mp 210–212 °C; yield 83%; UV max = 256 nm (ε = 16200); Rf = 0.25 in 1:1 hexanes–EtOAc; IR (cm−1): 3337, 3121, 2983, 2936, 2242 (CN), 1789, 1725 (CO), 1612 (CO), 1504, 1440, 1398, 1282, 1182, 1135, 1049, 899, 855, 808, 762, 733, 658, 559, 441; 1H NMR (300 MHz, 20 mg: 0.4 mL CD3CN): δ 2.88 (6H, s, CH3), δ 6.85 (s, NH), δ 8.62 (dd, ArH6, J 2, J 8), δ 8.70 (d, ArH5, J 8), δ 8.75 (d, ArH2, J 2); δ13C NMR (75 MHz, 20 mg: 0.4 mL CD3CN): δ 25.3 (s, CH3 × 2), δ 59.7 (s, hyd-C-5), δ 108.8 (s, C-4′), δ 116.5 (s, CN), δ 123.7 (q, CF3, J 136), δ 124.9 (q, C-2′, J 5), δ 130.4 (s, C-6′), δ 133.4 (q, C-3′, J 17), δ 137.0 (s, C-5′), δ 138.2 (s, C-1′), δ 154.4 (s, hyd-C-2), δ 177.1 (s, hyd-C-4); GC-(EI)TOF-HRMS: calcd for C13H10N3O2F3 (M): 297.0725, observed: m/z 297.0713.
4-[3-(4-Hydroxybutyl)-4,4-dimethyl-2,5-dioxoimidazolidin-1-yl]-2-(trifluoromethyl)benzonitrile (RU58841)
Anhydrous conditions were kept by oven drying of glassware and flame drying with a Schlenk line using the vac/purge method. Compound 6 (1.00 g, 3.37 mmol) was dissolved in dry DMF (25 mL). NaH 60% suspension in mineral oil (280 mg, 7.00 mmol) was twice washed in a sealed flask with dry n-hexane (5 mL) using a syringe. The solution of 6 in DMF was then added to the flask containing the NaH by pressure equalizing funnel. The two were stirred for 15 min until bubbles of H2 gas ceased. 4-Bromobutyl acetate (680 mg, 3.49 mmol) was then added by syringe through the addition funnel and washed in with a second 25 mL portion of dry DMF. The mixture was stirred and heated at 50 °C for 2 h. A pellet of NaOH (∼200 mg) was added, followed by deionized water (45 mL).The crude yield was 92–98% when the DMF and water was removed by evaporation but for a more pure product the flask contents were cooled overnight at 0 °C and the solids collected for an 80% yield of RU58841 (996 mg, 2.70 mmol). Following instructions from Battmann et al.4 the product was recrystallized from diisopropyl ether. We found RU58841 hard to dissolve even in a large volume of diisopropyl ether. The addition of a similar volume of n-heptane as an anti-solvent was necessary followed by overnight standing at 8 °C to crystallize.
Mp 71–72 °C; yield 80%; UV max = 261 nm (ε = 15100); Rf = 0.07 in 1:1 hexanes–EtOAc or 0.18 in 1:3 hexanes–EtOAc or 0.42 in EtOAc; IR (cm−1): 3392 (OH), 3133, 2944, 2876, 2234 (CN), 1774, 1719 (CO), 1612 (CO), 1505, 1438, 1413, 1377, 1312, 1179 & 1133 (C–F), 1051, 894, 837, 763, 675, 555; 1H NMR (300 MHz, 20 mg: 0.4 mL CDCl3): δ 1.52 (6H, s, CH3), δ 1.64 (2H, m, CH2-3), δ 1.82 (2H, m, CH2-2), δ 2.12 (1H, s, OH), δ 3.39 (2H, t, CH2-1, J 6), δ 3.68 (2H, t, CH2-4, J 6), δ 7.89 (d, ArH5, J 8), δ 7.98 (dd, ArH6, J 2, J 8), δ 8.13 (d, ArH2, J 2); δ13C NMR (75 MHz, 20 mg: 0.4 mL CDCl3): δ 23.7 (s, CH3 × 2), δ 26.4 (s, al-C-2), δ 30.0 (s, al-C-3), δ 40.4 (s, al-C-1), δ 62.2 (s, hyd-C-5), δ 62.3 (s, al-C-4), δ 108.4 (s, C-4′), δ 115.3 (s, CN), δ 122.3 (q, CF3, J 136), δ 123.3 (q, C-2′, J 5), δ 128.2 (s, C-6′), δ 133.8 (q, C-3′, J 17), δ 135.6 (s, C-5′), δ 136.8 (s, C-1′), δ 153.2 (s, hyd-C-2), δ 174.9 (s, hyd-C-4); GC-(EI)TOF-HRMS: calcd for C17H18N3O3F3 (M): 369.1300, observed: m/z 369.1294.
C, H, N: calcd for C17H18N3O3F3: 55.28, 4.91, 11.38. Found: 55.19, 4.84, 11.28.
Crystallography
1 (CCDC 894556) was obtained from a single crystal grown in methanol and shows the correct structure.2 (CCDC 904089) was obtained after crystallization in DMF with the addition of water. The crystal structure revealed a 1:1 co-crystallization of 2 with DMF.
3 (CCDC 893326) confirmed the structure. The experimental procedure gave a suitable crystal directly.
4 (CCDC 892388) this single crystal proved difficult to grow and involved first co-crystallizing with m-xylene, removal of solvent under high vacuum at 60 °C, then recrystallizing in toluene to give a single crystal for diffractometry.
Acknowledgements
We acknowledge the School of Applied Sciences and College of Science, Health & Engineering at RMIT for providing Matthew's PhD scholarship. We would like to thank Frank Antolasic at RMIT and Sally Duck at Monash University for help with mass spectrometry measurements. We also thank Jörg Wagler at Technische Universität Bergakademie, Freiberg for assistance with crystallography. We acknowledge Jamie Simpson at Monash Institute of Pharmaceutical Sciences for use of lab space. We thank Bob McAllister at University of Otago for C,H,N analyses.Notes and references
- C. Montagne and M. Shipman, Synlett, 2006, 2203–2206 CAS.
- P. Chemla, Tetrahedron Lett., 1993, 34(46), 7391–7394 CrossRef CAS.
- A. Volonterio and M. Zanda, Tetrahedron Lett., 2003, 44, 8549–8551 CrossRef CAS PubMed.
- T. Battmann, A. Bonfils, C. Branche, J. Humbert, F. Goubet, G. Teutsch and D. Philibert, J. Steroid Biochem. Mol. Biol., 1994, 48(1), 55–60 CrossRef CAS.
- D. Cousty-Berlin, B. Bergaud, M. C. Bruyant, T. Battmann, C. Branche and D. Philibert, J. Steroid Biochem. Mol. Biol., 1994, 51(1/2), 47–55 CrossRef CAS.
- S. H. Windahl, R. Galien, R. P. Chiusaroli, P. Clément-Lacroix, F. Morvan, L. N. Lepescheux, F. O. Nique, W. C. Horne, M. L. Resche-Rigon and R. Baron, J. Clin. Invest., 2006, 116(9), 2500–2509 CAS.
- T. Battmann, C. Branche, F. Bouchoux, E. Cerede, D. Philibert, F. Goubet, G. Teutsch and M. Gaillard-Kelly, J. Steroid Biochem. Mol. Biol., 1998, 64(1–2), 103–111 CrossRef CAS.
- B. De Brouweri, C. Tetelini, T. Leroyi, A. Bonfils and D. Van Neste, Br. J. Dermatol., 1997, 35(5), 699–701 CrossRef PubMed.
- W. Gao, C. Bohl and J. Dalton, Chem. Rev., 2005, 105, 3352–3370 CrossRef CAS PubMed.
- H. M. Hügel, C. J. Rix and K. Fleck, Synlett, 2006, 2290–2292 CrossRef PubMed.
- K. Fleck, PhD thesis, Synthetic approaches to the N-Arylation of imides, RMIT University, 2005, p. 101 Search PubMed.
- H. Sayo, H. Ohomori, T. Umeda and M. Masui, Bull. Chem. Soc. Jpn., 1971, 45, 203–208 CrossRef.
- M. B. Smith and J. March, March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, John Wiley & Sons, 6th edn, 2007, p. 571, (and references therein) Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 892388, 893326, 894556 and 904089. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra00332b |
| ‡ It is possible to capture 2-nitropropane as a commercial by-product during this step as we have observed it on a small scale reactive distillation. However optimal conditions for simultaneous ring closure and capture of 2-nitropropane have not yet been developed. |
| This journal is © The Royal Society of Chemistry 2014 |