PET depolymerisation in supercritical ethanol catalysed by [Bmim][BF4]†
Cátia Santos Nunes,
Michael Jackson Vieira da Silva,
Danielle Cristina da Silva,
Adonilson dos Reis Freitas,
Fernanda Andréia Rosa,
Adley Forti Rubira and
Edvani Curti Muniz*
Departamento de Química, Universidade Estadual de Maringá, Av. Colombo, 5790, 87020-900, Maringá-PR, Brazil. E-mail: ecmuniz@uem.br
First published on 15th April 2014
Abstract
Poly(ethylene terephthalate) (PET) was successfully depolymerised under supercritical ethanol. Robust conversion of 98 wt% from PET to diethylterephthalate (DET) was obtained by adding [Bmim][BF4], as catalyst, accompanied by reduction of depolymerization time from ca. 6 h to 45 min. DET formation in the depolymerization process was characterized by HPLC, 1H NMR, FTIR, TGA, DSC and SEM showing high purity and yield. The yields for different runs were determined by HPLC combined with interpolation from the standard/calibration curve. A 23 factorial design was employed to evaluate the effect of different inputs such as (i) reaction time after supercritical condition, (ii) volume of ionic liquid (VIL) and (iii) amount of PET in the yield of DET. By the analysis of variance (ANOVA), including F-test and P-values, it was found that reaction time and amount of PET inputs correspond, respectively, to 44% and 23% of the evaluated response. Another positive aspect showed by the factorial design is that the amount of catalyst was not significant in the process, and the depolymerization can be conducted successfully since a small amount (this study used VIL ranging 0.15 to 0.35 mL) is present in the reaction media. The method proposed in this paper is advantageous over others, reported in the literature, due to the lower reaction time required for PET depolymerization and the higher DET yield.
Introduction
The chemical recycling of polymers is an environmentally-friendly process, an alternative which adds value to polymer production waste or post-consumer polymer waste, where the product can be converted into its respective monomers and recycled for use in new polymerization products. Several depolymerisation techniques, especially those using catalytic reactions, have been studied extensively and the most studied catalysts are bases (ex. NaOH), acids (ex. H2SO4),1,2 and metal salts.3 Developments in catalysis are being constantly reported, trying to find new catalysts, novel catalytic reactions and alternative methodologies. Much of the pressure for this is driven by the economic requirements to develop systems in which the easy separation of products and/or the reuse of the catalyst are possible, along with desired high reactivity and selectivity.4 Chemical recycling of poly(ethylene terephthalate) (or PET) started in the 1950s at about the same time that PET was being manufactured on a commercial scale.5 (PET) is widely used in the manufacture of video and audio tapes, films for X-ray diagnostics, food packaging and soft-drink bottles.6Several processes for PET depolymerization have been put forward with different depolymerizing agents and operation conditions. Alcoholysis processes depolymerize PET to dimethyl terephthalate (DMT) with liquid or gaseous methanol7–9 glycolysis depolymerizes PET to bis-(hydroxyethyl) terephthalate (BHET) with ethylene glycol or other glycol,10,11 and hydrolysis1 converts PET to terephthalate acid (TPA) under the promotion of acidic or basic conditions. All of these processes have both advantages and disadvantages. Methanolysis under normal conditions can be carried out at relative mild temperature and pressure. However, the reaction rate is very slow, and some divalent metal catalysts such as zinc, lead, and manganese acetates, are required to enhance depolymerization rate. The undesired diethylene glycol, the dimer of ethylene glycol, is formed in the glycolysis. There are thus some problems in separation and purification of the product.12 Hydrolysis under acidic or basic conditions may cause corrosion and pollution problems,13 whilst both terephthalic acid formed in the reaction and the acidic catalysts favour the formation of diethylene glycol. Some new experimental methods have been introduced for the decomposition of PET. As an example, microwave irradiation was used as energy source in solvolysis of PET,14 and a high-pressure calorimeter has been used for measure the decomposition degree.15 In recent years, supercritical fluids are very attractive media for conducting chemical transformations, primarily because the solvent and transport properties of a single solution can be appreciably and continuously varied with relatively minor changes in either temperature and/or pressure to achieve supercritical conditions.16 Supercritical fluids such as water and alcohol are excellent reaction media for the depolymerisation of plastics by using sub and/or supercritical conditions, because the reaction can proceed rapidly and selectively under such conditions.17 For instance, Kamimura et al.18 demonstrated the decomposition of waste material composed of polyamide 6 (nylon-6) into valuable compounds such as methyl 6-hydroxycapronate and methyl 5-hexenoate, in a ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and yield of 80%, using methanol under supercritical conditions. Gutiérrez et al.19 proposed a process that combines green solvents and supercritical fluid extraction to recycle polystyrene wastes, with a reduced volume, almost completely free of solvent and without degradation.
:1 and yield of 80%, using methanol under supercritical conditions. Gutiérrez et al.19 proposed a process that combines green solvents and supercritical fluid extraction to recycle polystyrene wastes, with a reduced volume, almost completely free of solvent and without degradation.
Processes involving the depolymerisation of PET under supercritical conditions were first used in Japan in 1997, where PET was depolymerised using supercritical water20 or supercritical methanol.21 More recently, supercritical ethanol (scEtOH) was employed for depolymerising PET from multilayer packaging films, and diethyl terephthalate (DET) was obtained as the main product with high purity and an at 80% yield.22
Ethanolysis as a PET depolymerisation method is a good alternative for the recycling industry, especially in countries (Brazil, for instance) where ethanol from sugarcane is abundant and relatively inexpensive.23 Ionic liquids (ILs), considered “green solvents”,24,25 have attracted enormous research interest due to their unique features, including optimization of compound characteristics through a broad selection of anion and cation combinations, thermal stability, non-volatility, electrochemical stability, and low flammability.26 In the last decade, ILs have been widely used in extraction, catalysis, electrochemistry,27 organic synthesis28 and may have applications in polymer research.29 Growing interest has focused on ILs as catalysts, in either homogenous or heterogeneous media,24,25 combining the advantages of a solid for a surface-immobilizing catalyst and the advantages of a liquid for allowing the catalyst to move freely.30
In this study, [Bmim][BF4] was used as the catalyst for the depolymerisation of PET under scEtOH. The aim of this work was to determine the influence of reaction time, amount of PET and volume of [Bmim][BF4] in the feed on PET depolymerisation under scEtOH and whether this procedure had potential technological applications. This work combines two important alternatives for sustainable reactions: the supercritical environment and the ionic liquid.24,25 To our knowledge, no literature has been published focusing this specific issue, in spite recent publication anticipated these conditions as future perspective for sustained developing of polymer chemistry.24,25
Experimental
Chemicals
Anhydrous ethanol (99.5 GL purity) was supplied by Nuclear (Diadema – SP, Brazil). Diethylterephthalate (DET) and bis-(hydroxyethyl) terephthalate (BHET), used as standards, were purchased from Sigma-Aldrich (New Jersey, USA). Reagents used for synthesis of [Bmim][BF4] were N-methylimidazole (99%) (Sigma-Aldrich, St. Louis, MO, USA), 1-chlorobutane, acetonitrile and dichloromethane (Merck, Whitehouse Station, New Jersey, USA). Potassium tetrafluoroborate was acquired from Strem Chemicals Inc. (Newburyport, MA, USA). Acetonitrile (Merck, Whitehouse Station, New Jersey, USA) was distilled over phosphorus pentoxide (P2O5),31 1-chlorobutane (Whitehouse Station, New Jersey, USA) was used as received, ethyl acetate (Merck, Whitehouse Station, New Jersey, USA) was distilled over P2O5.31PET (pellets, average largest dimension ca. 1 mm) from waste soft drinks bottles (Plaspet Reciclagens Ltda company, Maringa, Brazil) was washed and dried in an oven at 50 °C to a constant weight. Viscosity measurements of PET (from the same source) at 25 °C in a 1:1 solution of 1,2-dichlorobenzene/phenol (w/w) estimate3 the average molecular weight viscosity of PET (Mv) as being 54600 g mol−1.
Equipments and procedures
FTIR spectra used to characterize the chemical structures of the depolymerisation products were recorded using a (Bomem model MB-100 spectrometer, Quebec, Canada) in the range of 4000–400 cm−1. To quantify the DET formed at the end of each depolymerisation run, RP-HPLC analysis was performed using a Thermo Surveyor LC Pump Plus, PDA Plus Detector set at 240 nm and a reverse-phase C18-Kromasil column (250 × 4.6 mm) with a particle size of 5 μm and an average pore size of 100 Å. A methanol/water 80/20 v/v mixture was used as the mobile phase at a flow rate of 1 mL min−1. Injection of 50 μL (after be degassed) and a photodiode detector with a 5 cm optic length were used. An analytical curve using standard DET solutions at concentrations ranging from 0.24 to 2.00 mg mL−1 was built. 1H NMR spectra were used to characterize the chemical structures of depolymerisation products and were obtained on a (Varian model Mercury Plus, 300 spectrometer, Palo Alto, CA, USA) operating at 300 MHz and calibrated with tetramethysilane (TMS) as an internal reference. Analyses of 1H NMR were performed according to the methods published by Castro et al.23 1H NMR spectrum of standard DET was obtained and used for comparison. Scanning electron microscope (SEM, Shimadzu, model SS550 Superscan, Japan), applying voltage of 15.0 kV and current of 30 mA, was used for morphology analyses. Before the SEM analyses, the DET obtained at the final of depolymerization reaction (run 8) was precipitated in water and further freeze dried. Finally, the treated DET sample was coated with a thin gold film and analyzed by SEM. SEM of standard DET was obtained and used for comparison.The thermal properties of raw PET and depolymerisation products were investigated by TGA and DSC. TGA experiments were carried out in a thermogravimetric analyzer (Netzsch, model STA 409 PG/4/G Luxx, New Castle, Delaware, USA) at 30 to 550 °C at a rate of 10 °C min−1 under N2 gas flowing at a rate of 20 mL min−1. DSC analyses were performed on a calorimeter (Netzsch, model STA 409 PG/4/G Luxx, New Castle, Delaware, USA) at a temperature range of 40–210 °C, heating rate of 10 °C min−1 and nitrogen flow of 50 mL min−1.
Synthesis of 1-n-butyl-3-methylimidazolium tetrafluoroborate ([Bmim][BF4])
The 1-butyl-3methylimidazolium chloride was synthesized according to the methods described by Dupont et al.31 For this, a 100 mL, three-necked, round-bottomed flask equipped with a heating oil bath, a nitrogen inlet adapter, an internal thermometer adapter, an overhead mechanical stirrer, and a reflux condenser were used. The flask was flushed with nitrogen and charged with 5 g (0.06 mol) of freshly distilled N-methylimidazole, 5 mL of acetonitrile (CH3CN) and 7.22 g (0.078 mol) of 1-chlorobutane, brought to a gentle reflux (75–80 °C, internal temperature), heated under reflux for 48 h and cooled to room temperature. The volatile material was removed from the resulting yellow solution under reduced pressure. The remaining light-yellow oil was re-dissolved in dry acetonitrile (8.1 mL) and added drop-wise via cannula to 30 mL of a well-stirred solution of dry ethyl acetate and one seed crystal of 1-butyl-3-methylimidazolium chloride placed in a 100 mL, three-necked, round-bottomed flask equipped with a nitrogen inlet adapter and an overhead mechanical stirrer. The imidazolium salt began to crystallize exothermically almost immediately, and after addition of the acetonitrile solution had been completed, the flask was cooled at −30 °C for 2 h. The supernatant solution was removed via filtration through a filter cannula and the resulting white solid dried under reduced pressure (0.1 mbar) at 30 °C for 6 h to yield 9.33 g (89%) of 1-butyl-3-methylimidazolium chloride ([Bmim][Cl]) having a melting point (mp) of 66–67 °C.The [Bmim][BF4] was synthesized according to the methods outlined by Dupont et al.31 using the previously prepared [Bmim][Cl]. For this, a 100 mL, one-necked, round-bottomed flask was charged with 5 g (0.028 mol) of finely powdered [Bmim][Cl] and 3.6 g (0.028 mol) of potassium tetrafluoroborate in 10 mL of distilled water. The system was stirred at room temperature for 2 h yielding a heterogeneous mixture and the water removed under reduced pressure (0.1 mbar) at 80 °C until a constant weight was reached. In sequence, 6 mL of dichloromethane and 1.88 g of anhydrous magnesium sulphate were added to the remaining suspension. After 1 h, the suspension was filtered and the volatile material removed under reduced pressure (0.1 bar) at 30 °C for 2 h to yield 5.65 g (0.025 mol, 91%) of 1-butyl-3-methylimidazolium tetrafluoroborate as a light yellow, viscous liquid having a mp of −74 °C.
Depolymerisation of PET under scEtOH catalysed by [Bmim][BF4]
The process was performed according to the methodology14 using the same apparatus. The degradation reaction was carried out in a 0.1 L home-made batch-type reactor made of 316® stainless steel equipped with inlet and outlet valves, a manometer, a thermometer, and a heating collar controlled by a programmable temperature controller used to heat the reactor to the desired temperature (255 °C, with a precision estimated to be 5 °C, measured by a J-type thermocouple). For the experiments, the desired mass of PET, anhydrous ethanol and [Bmim][BF4] were added to the reactor at room temperature according to a 23 factorial design (see Table 1) and heated (at heating rate of about 8 °C min−1) to a reaction temperature of 255 °C. Reactor pressure (115 atm) was attained by nearly filling the vessel with desired amount of PET (pellets) and ethanol and keeping the reactor properly closed, avoiding the need of using pressurized gas for the ethanol to reach supercritical conditions. After a required reaction time, the heating collar was removed and the vessel quickly cooled to room temperature using large amounts of water, taking less than 5 min to reach room temperature (around 25 °C).| Runs | Inputs | Yield (wt%) | ||
|---|---|---|---|---|
| RT | VILs | WPET | ||
| 1 | − | − | − | 34.6 |
| 2 | + | − | − | 64.0 |
| 3 | − | + | − | 13.8 |
| 4 | + | + | − | 91.6 |
| 5 | − | − | + | 66.0 |
| 6 | + | − | + | 69.2 |
| 7 | − | + | + | 71.3 |
| 8 | + | + | + | 98.0 |
Recovering of the ionic liquid
After PET despolymerization (run 8) ethanol was evaporated and the main product (DET) was precipitated in the water and filtered. The residual ionic liquid [Bmim][BF4] remaining in the filtrate aqueous was lyophilized prior the 1 HNMR analyses. The spectrum were obtained as earlier described and presented in ESI (Fig. S1†).Factorial design experiments
The influences of reaction time (RT), amount of PET (WPET) and ionic liquid volume (VILs) used for PET depolymerisation by scEtOH on the yield of DET were investigated. A two level factorial (23) design for evaluating each of these parameters was used [RT: (+) 45 min, (−) 0 min; WPET: (+) 1.5 g, (−) 0.5 g and VILs: (+) 0.35 mL, (−) 0.15 mL] and DET yield (in wt%) was analysed (see Table 1) as response. A linear model was applied to fit the experimental data. The response surface, produced after applying a given linear model to the data, was obtained and the respective analysis of variance (ANOVA) as performed using the (Design Expert DX7© software Version 7.0, Minneapolis, MN, USA). The sequence of runs was random.Results and discussion
Products obtained from the depolymerisation of PET under scEtOH catalysed by [Bmim][BF4]
The stability of [Bmim][BF4] under supercritical ethanol (scEtOH) used for PET depolymerisation was analysed. FTIR and 1H NMR spectra of [Bmim][BF4], before and after the ionic liquid (IL) exposure to 115 atm and 255 °C for 60 min, were obtained. No changes were observed in the [Bmim][BF4] FTIR and 1 HNMR spectra (Fig. S2 and S3, ESI†) after exposure, which indicated [Bmim][BF4] was chemically stable when exposed to these T and P conditions for 60 min.The products obtained from PET depolymerisation under scEtOH were primarily DET and either ethylene glycol (EG), mono-(hydroxyethyl) terephthalate (MHET) and bis-(hydroxylethyl) terephthalate (BHET) with the possibility of other by-products like ethylene terephthalate (ET), dimers and oligomers. These data verified the reaction in which DET and EG were formed as the main products (Scheme 1).32 For that, each PET ester linkage used one ethanol molecule in the depolymerisation process, creating two ethyl end-groups to form one DET molecule and yielding two diethyleneglycol (EG) molecules for each DET molecule, while BHET was formed by further reaction between DET and EG.
 | ||
| Scheme 1 Proposed mechanism for PET depolymerisation under scEtOH in the presence of the ionic liquid [Bmim][BF4]. Adapted from ref. 32. | ||
The proposed mechanism for the PET depolymerisation reaction investigated in the present work is shown in Scheme 1, based in a paper published by Liu et al.32
Influence of reaction time (RT), VILs and WPET on PET depolymerisation under scEtOH
The influence of [Bmim][BF4] in the chemical recycling of the PET under supercritical ethanol (T = 255 °C and P = 115 atm) was investigated through a 23 factorial design. Table 1 shows the conditions used in each of the eight runs and Table 2 shows the ANOVA obtained after treating the data collected from such factorial design. The effects of reaction time (RT), ionic liquid volume (VILs) and amount of PET (WPET) inputs, their second order interactions on DET yield, and the respective F-ratio values are presented in Table 2.| Source of variation | Sum of squares | Degree of freedom | Mean square | F value | P value |
|---|---|---|---|---|---|
| Model | 5214.30 | 5 | 1042.86 | 12.22 | 0.0774 |
| A: RT | 2349.55 | 1 | 2349.55 | 27.53 | 0.0345 |
| B: LIs | 209.10 | 1 | 209.10 | 2.45 | 0.2580 |
| C: WPET | 1262.53 | 1 | 1262.53 | 14.80 | 0.0614 |
| AB | 646.20 | 1 | 646.20 | 7.57 | 0.1106 |
| AC | 746.91 | 1 | 746.91 | 8.75 | 0.0978 |
| Residual | 170.66 | 2 | 85.33 | ||
| Total correl. | 5384.96 | 7 |
After applying the linear model: yield = 63.56 + 17.14 (RT) + 5.11 (VILs) + 12.56 (WPET) + 8.99 RT VILs − 9.66 (RT WPET) to the data, the R2 coefficient value was 0.968 obtained dividing the sum of squares for the model (5214.30) by the sum of squares for the total correlation (5384.96). The R2 value indicated a good fit of the model to the experimental data. The analysis of variance (ANOVA) was also used to estimate the significance of the main and interaction effects of input in the response (DET yield). The mean squares in Table 2 were used to estimate the F value (F-test) for each parameter (model, inputs and input interactions) at 95% confidence.
Analyses of data presented in Table 2 suggested that RT had the greatest effect on PET depolymerisation, followed by WPET in the feed. VILs did not exert significant effect on DET yield (see the value of F-test on Table 2) when taking into account the range for this input (0.15 to 0.35 mL). Statistically significant interactions between RT and VILs and also between WPET and VILs were found, as DET yield increased by simultaneously changes the inputs to same direction (up or down). In addition, the response surface shown in Fig. 1 indicated that the amount of IL (VILs) itself was not statistically significant input but its importance to DET yield during depolymerisation of PET by scEtOH was due to its interaction with RT and WPET, an inherent characteristic of catalysts for a given chemical reaction.
 | ||
| Fig. 1 Response surface for WPET and RT inputs after applying the linear model to the data collected from the eight runs, as described in Table 1. | ||
When WPET, RT and VILs were maintained at higher levels (WPET = 1.5 g, RT = 45 min; VILs = 0.350 mL), DET was formed at a yield of 98 wt%. According to Castro et al.23 using scEtOH without ionic liquid the reaction yielded 66 wt% after 5 hours of reaction, while using the ionic liquid at 120–200 °C at room pressure,29 a complete depolymerisation of PET occurred after 6–10 h. Therefore, the combination of scEtOH and ionic liquid, as used for PET depolymerisation in this study, enabled shorter times for near-complete PET depolymerisation with higher DET yields and represents a good and sustained strategy for depolymerisation. So as stated in literature, the use of ionic liquids and/or supercritical conditions are important for polymer synthesis.24,25 This work shows that it is also important for PET depolymerisation reactions. It is shown in this paper that the combination of scEtOH and IL lead to almost complete depolymerisation of PET in short reaction times (less than 1 hour). This can be in future an alternative for depolymerization of PET contributing to solve environmental concerns.
When WPET, RT and VILs were maintained at lower levels (WPET = 0.5 g, RT = 0 min, VILs = 0.150 mL), (or run 1, Table 1), higher amounts of BHET were formed (as compared to run 8 in which such inputs were maintained at higher levels). HPLC chromatograms of the products obtained during PET depolymerisation (run 1, RT = 0 min; run 8, RT = 45 min) are shown in Fig. 2.
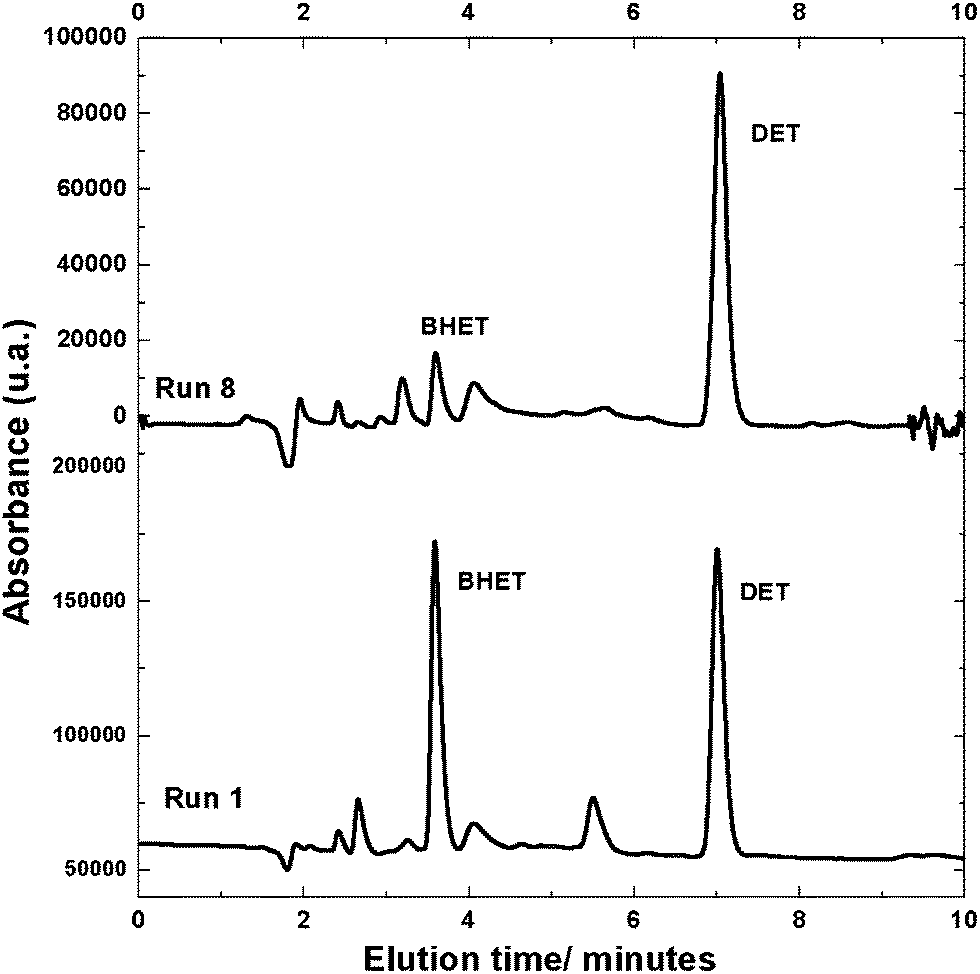 | ||
| Fig. 2 HPLC chromatograms of products obtained after PET depolymerisation under scEtOH from runs 1 and 8; conditions described in Table 1. | ||
The results in Fig. 2 indicated that DET, probably formed at the beginning of the PET depolymerisation reaction under scEtOH, further reacted with EG producing BHET. It should be noted that RT value was established as the period of time in which the system achieved scEtOH condition (T = 255 °C and P = 115 atm). In other words, the RT commenced when the system achieved the appropriate T and P condition, ca. 30 min after the heating apparatus was turned on. Therefore, RT equal to 0 min (run 1) indicated that the system had not achieved scEtOH condition, rather subcritical ethanol conditions, and BHET was formed in higher amounts in this condition (Fig. 2). This suggested that BHET should be more thermodynamically stable than DET, as the amount of BHET formed was higher at subcritical, rather than at supercritical ethanol conditions. However, for longer reaction times under scEtOH condition, DET was formed in greater amounts due to the presence of larger amounts of ethanol compared to EG, which pushed the equilibrium towards DET. These data indicated that the amount of BHET and/or DET formed during PET depolymerisation under scEtOH can be tuned by controlling temperature, pressure and reaction time.
Activity of [Bmim][BF4] during PET depolymerisation
[Bmim][BF4] showed significant activity during chemical depolymerisation by scEtOH. According to Garcia-Miaja et al.33 in a mixture of ethanol and [Bmim][BF4], BF4 anion is poorly capable of cross-associating with alcohol molecules, preferring to auto-associate, which is due to an increased system entropy (positive ΔSm). The effect of ionic liquids (ILs) in creating and/or altering the pathway of a given chemical reaction could be observed in reactions involving polar or electrically-charged intermediates such as carbocations and carbanions. From a synthetic chemistry point of view, the major benefit in the use ILs is the elevation of the reaction rate and selectivity related to others solvents.34 It can be pointed out that elevation of the reaction rate resulted from a decrease in free energy due to the presence of ILs that enables the formation of complexes that are more stable and have longer lifetimes in that media.35 Despite the complete understanding of the catalytic mechanism of ionic liquids being in its infancy,32 it was possible to predict/infer the mechanisms by which ILs such as [Bmim][BF4] acts.Ionic liquids are composed only of ions and the ionic nature of ILs can affect the course of the chemical reactions.34 Published data suggest that the strong electrostatic field possessed by dialkyl imidazolium cations, mediated by counterions, may play an important role in initiating the desired reaction.34 Liu et al.32,36 developed an IL ([Bmim][Ac]) that acts as a catalyst for the depolymerisation of polycarbonate (PC) in the presence of methanol, and the effects of temperature, reaction time, methanol and [Bmim][Ac] concentration on the methanolysis were examined. The authors show that the conversion of PC into its respective monomers was nearly 100%, with a 95 wt% yield of bisphenol A (BPA) formed at ambient pressure and 90 °C over a period of 2.5 h. Zhou et al.37 obtained ca. 71 wt% BHET yield during PET depolymerisation by the IL 1,3-diethylimidazolium triaceticzincate ([Deim][Zn(OAc)3]) at 180 °C and under ambient pressure.
Other runs performed in this study (under conditions not included in Table 1) showed that the presence of water in the system negatively affected [Bmim][BF4]-catalysed PET depolymerisation under scEtOH. For instance, using the conditions described for run 8, but in the presence of water at 1, 2, and 4 wt% (relative to amount of ethanol), DET yield decreased to 28.4, 13.8 and 13.8 wt%, respectively. The presence of water may have altered the interactions between the counterions of [Bmim][BF4], resulting in decreased catalytic activity. According to Khupse and Kumar,38 the presence of water, even in small amounts, decreases the viscosity of [Bmim][BF4] in hydrophilic and hydrophobic solvents, affecting counterion mobility and allowing dissociation into more free cations and anions. In this work, this could be responsible for the decreased catalytic effect of [Bmim][BF4] during PET depolymerisation under scEtOH in presence of low amounts of water.
According to literature, supercritical fluids, specifically supercritical water (Tc = 674.3 K, Pc = 22.0 MPa)20 and supercritical methanol (Tc = 512.3 K, Pc = 8.09 MPa)21 have been used for PET depolymerization. PET hydrolysis with supercritical water has very high reaction rate. But, in practice, this process is not easy to operate due to the severe reaction conditions (above 670 K, 30 MPa). In addition, the hydrolysis leads to low yield of ethylene glycol (about 20%). Comparing to supercritical hydrolysis, the supercritical methanolysis21 operated at relatively mild conditions. In same direction, ethanolysis23 is also easy to operate. The process described in this work combines the IL and supercritical ethanol for PET depolymerization is a novelty and an alternative for PET recovering.
Characterization of the main product
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O, respectively.
O and C–O, respectively.
 | ||
| Fig. 3 FTIR spectra of raw PET, DET (the main product of the depolymerisation reaction, runs 1 and 8) and standard DET. | ||
 | ||
| Fig. 4 1H NMR spectra of: (a) product from run 1; (b) product from run 8; and (c) 1H NMR spectra of DET from run 8 and standard DET. | ||
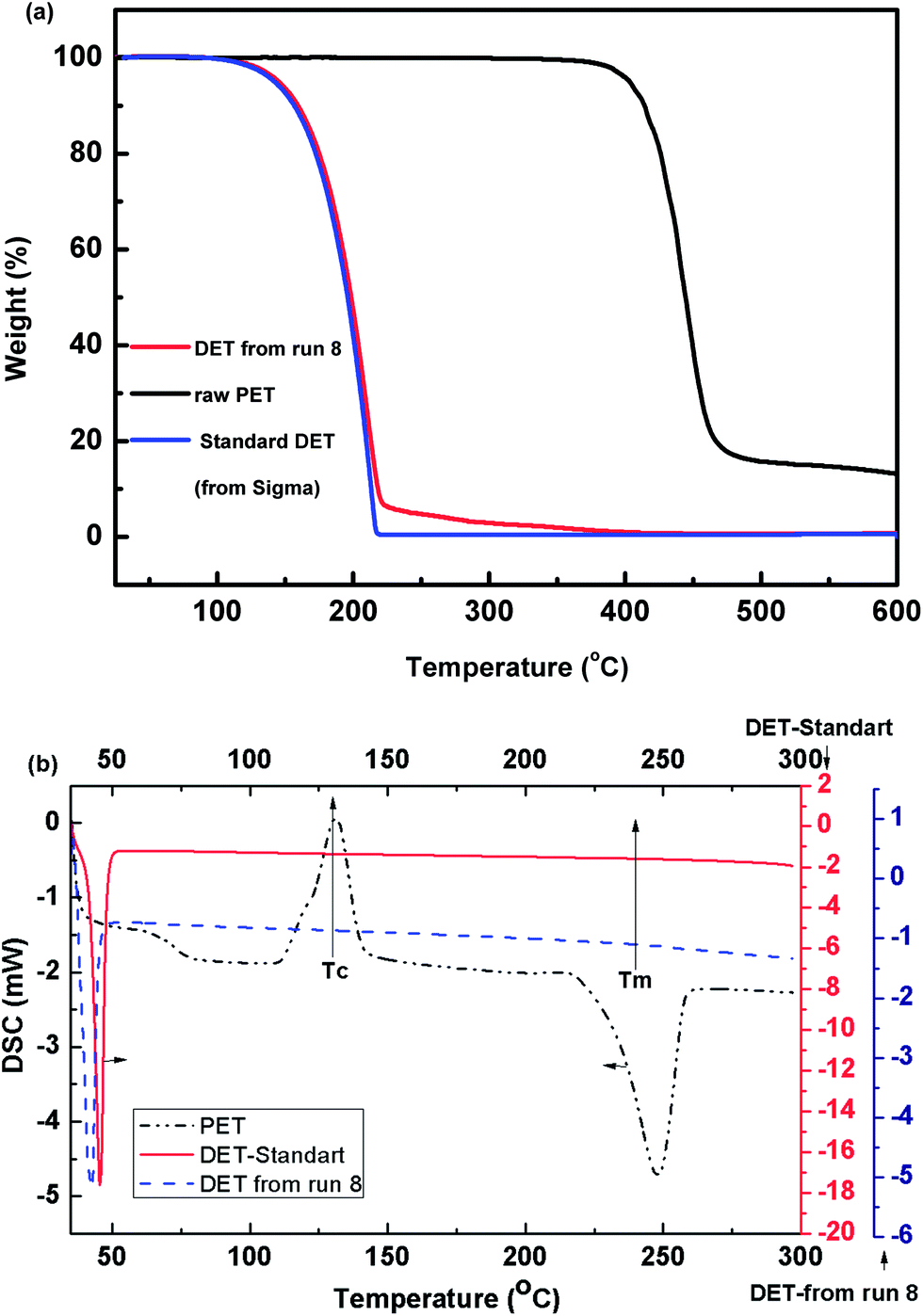 | ||
| Fig. 5 TGA (a); and DSC (b) curves of: raw PET, main product obtained from run 8, and the standard DET. | ||
The DSC curve of raw PET showed an endothermic peak at ∼235 °C related to the melting of raw PET,12 and an exothermic peak at ∼132 °C attributed to crystallization of PET. The intensity of peak attributed to crystallization is sensitive to ageing39 and to the thermal history40 of the PET sample. DSC of the product obtained from run 8 (DET) exhibited only an endothermic event that was attributed to the melting process. Comparing DSC and TGA curves of DET from run 8 to DSC and TGA curves of the standard DET suggested that the thermal properties of the product obtained from run 8 was quite similar to that of the standard DET. The minor differences in the DSC and TGA curves were attributed to the small fraction of BHET and MHET that are present in the sample as discussed earlier.
 | ||
| Fig. 6 SEM images of: DET from run 8 (a and b); and standard DET. | ||
Conclusions
The depolymerisation of PET under supercritical ethanol (scEtOH) in presence of the ionic liquid [Bmim][ BF4] was conducted, primarily yielding mainly diethyl terephthalate (DET) for longer reaction times (45 min). Physical and chemical characterization of the main products were performed by FTIR, 1H NMR, HPLC, TGA/DSC and SEM. Compared to results obtained for PET depolymerisation under scEtOH but in absence of an ionic liquid, the addition of [Bmim][BF4] in the reaction vessel resulted in a faster reaction rate and the yield of DET approached 100 wt%. The use of ionic liquids, in addition to supercritical ethanol, represented an extremely promising combination for PET depolymerisation in a sustained way. Further studies using this system addressing the reuse of [Bmim][BF4], understanding the effects of water and whether PET particle size has an influence on DET yield during depolymerisation under scEtOH is going on in our lab.Acknowledgements
The authors would like to thank CAPES/Brazil, for the doctoral fellowship (CNS) and CNPq/Brazil for financial support (Grant number 309005/2009-4).References
- T. Yoshioka, T. Sato and A. Okuwaki, J. Appl. Polym. Sci., 1994, 52, 1353–1355 CrossRef CAS.
- M. Z. S. D. Mancini, Polímeros, 2002, 12, 34–40 CrossRef PubMed.
- M. E. Viana, A. Riul, G. M. Carvalho, A. F. Rubira and E. C. Muniz, Chem. Eng. J., 2011, 173, 210–219 CrossRef CAS PubMed.
- C. M. Gordon, Appl. Catal., A, 2001, 222, 101–117 CrossRef CAS.
- G. P. Karayannidis, D. S. Achilias, I. D. Sideridou and D. N. Bikiaris, Eur. Polym. J., 2005, 41, 201–210 CrossRef CAS PubMed.
- D. Paszun and T. Spychaj, Ind. Eng. Chem. Res., 1997, 36, 1373–1383 CrossRef CAS.
- Process for the recovery of dimethyl terephthalate from polyethylene terephthalate, US Pat., 3321510, 1967.
- Recovery of terephthalic acid from waste or used prods of polyalkylene terephthalate polymers – comprises subjecting waste prods consecutively to methanolysis with superheated methanol vapours and hydrolysis with water (vapour) at high temps., Eur Pat., 662446, 1995.
- Separation of dimethyl terephthalate and ethylene glycol from residues from rectification of waste ethylene glycol, PL Pat., 126009, 1985.
- J. Y. Chen, C. F. Ou, Y. C. Hu and C. C. Lin, J. Appl. Polym. Sci., 1991, 42, 1501–1507 CrossRef CAS.
- S. Baliga and W. T. Wong, J. Polym. Sci., Part A: Polym. Chem., 1989, 27, 2071–2082 CrossRef CAS.
- M. Ghaemy and K. Mossaddegh, Polym. Degrad. Stab., 2005, 90, 570–576 CrossRef CAS PubMed.
- C. Y. Kao, W. H. Cheng and B. Z. Wan, J. Appl. Polym. Sci., 1998, 70, 1939–1945 CrossRef CAS.
- A. Krzan, J. Appl. Polym. Sci., 1998, 69, 1115–1118 CrossRef CAS.
- Y. Kishimoto, T. Kajihara and S. Kato, Polym. Bull., 1999, 42, 295–300 CrossRef CAS.
- K. W. Hutchenson, in Supercritical Fluid Technology in Material Science and Engineering, New York, 2002 Search PubMed.
- M. Goto, J. Supercrit. Fluids, 2009, 47, 500–507 CrossRef CAS PubMed.
- A. Kamimura, K. Kaiso, S. Suzuki, Y. Oishi, Y. Ohara, T. Sugimoto, K. Kashiwagi and M. Yoshimoto, Green Chem., 2011, 13, 2055–2061 RSC.
- C. Gutierrez, M. T. Garcia, I. Gracia, A. de Lucas and J. F. Rodriguez, J. Mater. Cycles Waste Manage., 2012, 14, 308–316 RSC.
- Y. Yang, Y. J. Lu, H. W. Xiang, Y. Y. Xu and Y. W. Li, Polym. Degrad. Stab., 2002, 75, 185–191 CrossRef CAS.
- T. Sako, T. Sugeta, K. Otake, N. Nakazawa, M. Sato, K. Namiki and M. Tsugumi, J. Chem. Eng. Jpn., 1997, 30, 342–346 CrossRef CAS.
- S. L. Favaro, A. R. Freitas, T. A. Ganzerli, A. G. B. Pereira, A. L. Cardozo, O. Baron, E. C. Muniz, E. M. Girotto and E. Radovanovic, J. Supercrit. Fluids, 2013, 75, 138–143 CrossRef CAS PubMed.
- R. E. N. de Castro, G. J. Vidotti, A. F. Rubira and E. C. Muniz, J. Appl. Polym. Sci., 2006, 101, 2009–2016 CrossRef CAS.
- K. Schroder, K. Matyjaszewski, K. J. T. Noonan and R. T. Mathers, Green Chem., 2014, 16, 1673–1686 RSC.
- P. Pollet, E. A. Davey, E. E. Urena-Benavides, C. A. Eckert and C. L. Liotta, Green Chem., 2014, 16, 1034–1055 RSC.
- M. Kosmulski, J. Gustafsson and J. B. Rosenholm, Thermochim. Acta, 2004, 412, 47–53 CrossRef CAS PubMed.
- M. Galinski, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567–5580 CrossRef CAS PubMed.
- H. Hagiwara, Y. Sugawara, K. Isobe, T. Hoshi and T. Suzuki, Org. Lett., 2004, 6, 2325–2328 CrossRef CAS PubMed.
- H. Wang, Z. Li, Y. Liu, X. Zhang and S. Zhang, Green Chem., 2009, 11, 1568–1575 RSC.
- R. Morland, American Chemical Society Division of Industrial and Engineering Chemistry, San Diego, 2001 Search PubMed.
- C. S. C. J. Dupont, P. A. Z. Suarez and R. F. de souza, Org. Synt. Coll., 2004, 10, 184 Search PubMed; C. S. C. J. Dupont, P. A. Z. Suarez and R. F. de souza, Org. Synt. Coll., 2002, 79, 236 CrossRef.
- F. Liu, L. Li, S. Yu, Z. Lv and X. Ge, J. Hazard. Mater., 2011, 189, 249–254 CrossRef CAS PubMed.
- G. Garcia-Miaja, J. Troncoso and L. Romani, Fluid Phase Equilib., 2008, 274, 59–67 CrossRef CAS PubMed.
- M. A. P. Martins, C. P. Frizzo, D. N. Moreira, N. Zanatta and H. G. Bonacorso, Chem. Rev., 2008, 108, 2015–2050 CrossRef CAS PubMed.
- Z. Y. Duan, Y. L. Gu and Y. Q. Deng, J. Mol. Catal. A: Chem., 2006, 246, 70–75 CrossRef CAS PubMed.
- F. Liu, Z. Li, S. Yu, X. Cui and X. Ge, J. Hazard. Mater., 2010, 174, 872–875 CrossRef CAS PubMed.
- X. Zhou, X. Lu, Q. Wang, M. Zhu and Z. Li, Pure Appl. Chem., 2012, 84, 789–801 CrossRef CAS.
- N. D. Khupse and A. Kumar, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2010, 49, 635–648 Search PubMed.
- E. A. McGonigle, J. H. Daly, S. Gallagher, S. D. Jenkins, J. J. Liggat, I. Olsson and R. A. Pethrick, Polymer, 1999, 40, 4977–4982 CrossRef CAS.
- W. Dong, J. Zhao, C. X. Li, M. L. Gu, D. L. Zhao and Q. R. Fan, Polym. Bull., 2002, 49, 197–203 CrossRef CAS PubMed.
- H. Wang, Y. Q. Liu, Z. X. Li, X. P. Zhang, S. J. Zhang and Y. Q. Zhang, Eur. Polym. J., 2009, 45, 1535–1544 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra00262h |
| This journal is © The Royal Society of Chemistry 2014 |