
Electronic effects of para substituents of 2-hydroxybenzaldehyde co-ligands in a family of hydrazonato-oxidovanadium(v) complexes†
Abstract
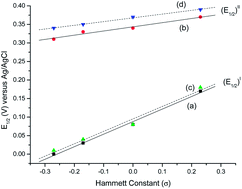
Eight mixed-ligand hydrazonato-oxidovanadium(V) complexes of the type [VVO(HL1)(A1–4)] (1–4) and [VVO(HL2)(A1–4)] (5–8) have been investigated, where (HL1)2− and (HL2)2− represent the dianionic forms of the 2-hydroxybenzoylhydrazone of acetylacetone (H3L1) and benzoylacetone (H3L2) respectively, and (A1–4)− represents the monoanionic forms of 2-hydroxybenzaldehyde (HA1), 2-hydroxy-5-methylbenzaldehyde (HA2), 2-hydroxy-5-methoxybenzaldehyde (HA3) and 5-bromo-2-hydroxybenzaldehyde (HA4) respectively. The complexes were synthesized by refluxing [VIVO(acac)2] and [VIVO(bzac)2], where acac− and bzac− are the monoanionic forms of acetylacetone (Hacac) and benzoylacetone (Hbzac) respectively with an equimolar amount of 2-hydroxybenzoylhydrazide in methanol followed by the addition of either an equivalent amount or a slight excess of each HA. 1H NMR spectra of complexes 1–3 and 5–7 in CDCl3 indicated the existence of two isomeric forms arising from the interchange of the coordinating positions of the bidentate A− ligand between axial and equatorial positions, a conclusion which is also supported by 51V NMR spectra. The vanadium atom in these complexes is in a six-coordinate distorted octahedral environment in which the dinegative hydrazone ligands in the enol form bind the vanadium in a tridentate meridional fashion utilizing their enolic-O, one imine-N and amide-O atoms, leaving the phenolic-O and other imine-N atoms unbonded although they do form intramolecular hydrogen bonds as is evident from IR, 1H NMR and X-ray diffraction studies. Analysis of electronic spectra and redox potential (E1/2) values of these complexes indicate that the λmax values for the LMCT transition and the E1/2 values are linearly related to the Hammett σ constants of the substituents in the monoanionic aryloxy ring of the A− co-ligand. On keeping in methanol for ∼5 days, the monomers 1–8 are converted to dimethoxide bridged dimeric [VVO(HL)(OCH3)]2 (9 and 10) complexes while oxido bridged dimeric [VVO(HL)]2O (11 and 12) complexes are obtained from dichloromethane or chloroform solution at room temperature after ∼5 days. On dissolution in CHCl3 or CH2Cl2, the [VVO(HL)(OCH3)]2 complexes converted to their respective monomeric [VVO(HL)(OCH3)] analogues and also to oxido bridged dimeric [VVO(HL)]2O species which is evident from their 1H NMR, 51V NMR and mass spectra.
 Please wait while we load your content...
Please wait while we load your content...

