A cyanide selective off–on fluorescent chemosensor with in vivo imaging in 100% water: solid probe preferred over in situ generation†
Sanju Dasab,
Surajit Biswasa,
Santanu Mukherjeec,
Jaya Bandyopadhyayc,
Subhodip Samantab,
Indrani Bhowmickd,
Dipak Kumar Hazrae,
Ambarish Ray*b and
Partha Pratim Parui*a
aDepartment of Chemistry, Jadavpur University, Kolkata 700032, India. E-mail: parthaparui@yahoo.com; Fax: +91-33-24146223; Tel: +91-9433490492
bDepartment of Chemistry, Maulana Azad College, Kolakta 700013, India. E-mail: r_ambarish@yahoo.co.in; Fax: +91-33-22268111; Tel: +91-9836650180
cDepartment of Biotechnology, West Bengal University of Technology, Kolkata 700064, India
dDepartment of Chemistry, University of Delhi, Delhi 110007, India
eDepartment of Solid State Physics, Indian Association for the Cultivation of Science, Kolkata 700032, India
First published on 28th January 2014
Abstract
A nontoxic fluorescent chemosensor [Cu(BP)HMB]2(ClO4)2 (1) synthesized in solid phase, exhibits unprecedented selectivity and sensitivity over the allied in situ complexes to perform fluorescence in “turn-off–on” mode for sensing cyanide in 100% aqueous medium under physiological conditions and for in vivo imaging using the nematode C. elegans. Below μM detection limit, instantaneous and excellent ratiometric responses are also beneficial to detect trace amounts of anthropogenic or biogenic cyanide.
The design and development of chemosensors capable of detecting selective toxic and lethal anionic species are of current research interest in chemistry, biology, medicine and in relation to environmental issues.1 Among various biologically hazardous anions, cyanide (CN−) is considered to be the most potent one.2 According to the World Health Organization (WHO), the maximum acceptable level of CN− in drinking water is 1.9 μM.3 However, the widespread use of CN− in industries and their waste effluents impose serious threat to the aquatic environment.4 Hence, intensive effort should be given for the development of 100% water soluble selective and sensitive CN− chemosensor with rapid response for effective in vivo detection. The detection principles of such chemosensors are based on mainly H-bonded/self-assembled receptor approach,1b,c,5 displacement approach1a,5a,6 or chemidosimeter approach,5a,7 although majority of them are operative in organic solvents or mixed aqueous solvents and sometimes not even applicable for liquid phase at all.1,5–7 Considering the advantage–disadvantage parameters of those approaches, it is presumed that the transition metal based cationic fluorescent chemosensors by displacement approach would be the optimal choice to maintain aforesaid all criteria for CN− detection in aqueous medium.1a,5a,6 However, discovery of completely water soluble sensing probe with sensitivity to detect below μM CN− is still a limiting phenomenon.5b,6a
In Cu(II) mediated displacement approach, a fluorescent probe is designed based on a giant organic dye whose fluorogenic activity is totally quenched by the complexation with Cu(II). On exposure to CN−, decomplexation occurs to form stable [Cu(CN)x]n− species and fluorogenic activity reappears from the liberated dye.1a,5a Unfortunately, owing to scarce solubility of those dyes and their copper complexes in aqueous medium, possibilities of in vivo application are relatively obscure. Generally, toxic organic solvents in different extents are used to solubilize the sensors in aqueous medium, except one rare report is there which operates in 100% aqueous medium with in vivo application.6a Again, in most of the cases, sensors were synthesized under in situ condition without solid state isolation.5–7 Hence the detail analytical comparisons in terms of efficiency, in between the solid probe and that generated in situ, are still missing in the literatures.
Herein, we report the design and synthesis along with detailed structural analysis of a solid fluorescent chemosensor [Cu(BP)HMB]2(ClO4)2 (1) (BP = 2, 2′-bipyridine), based on a tiny organic chromophore, 2-hydroxy-3-(hydroxymethyl)-5-methylbenzaldehyde (HHMB), operating in “turn-off–on” mode with high selectivity and sensitivity for recognizing the CN− in 100% aqueous medium as well as in living biological system. It is noteworthy that in aqueous medium, the dimeric solid probe (1) undergoes irreversible change to its monomeric form [Cu(BP)(HMB)(H2O)2](ClO4) (1a) which is actually responsible for cyanide attack (vide infra) as depicted in Scheme 1. The water soluble organic compound HHMB was prepared according to the similar method reported previously with some additional modifications8 (ESI†). It behaves as an organic fluorophore with a moderately high quantum yield (ϕF) ca. 0.05 in aqueous medium (ESI†). The extent of fluorescence quenching of HHMB was determined for in situ complexation with Cu(ClO4)2, where inadequate quenching in presence of saturating 6-equivalent Cu(II) ions precludes its selection to act as the sensor in “turn-off” mode. Interestingly, under similar condition, the fluorogenic activity of HHMB was quenched quite efficiently by strategically attaching the strong field ligand BP in Cu(II) precursor as Cu(BP)(ClO4)2. The strong field character of the BP may enhance the electron transfer process more facile for sufficient quenching of HHMB fluorescence to perform appropriate “turn-off” mode of sensing (Fig. S1, ESI†). Meanwhile, in absorption titrations, the depletion of 340 nm absorption band of HHMB with concomitant formation of a new absorption band ca. 400 nm in both cases with the isobestic point ca. 367 nm were due to ligand to Cu(II) charge transfer (CT) during complexation (Fig. S2, ESI†). We are fortunate enough to synthesize the more preferred sensor 1 in solid state and characterized the structure by the single crystal X-ray diffraction, where the sensor 1 crystallizes in the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with Z = 2 to exist as dimer in solid phase (Fig. 1) (ESI†). In aqueous medium, the dimeric core probably breaks up into two monomeric units. The most probable monomeric structure (1a) in aqueous solution was optimized in gas phase by DFT calculation using Gaussian 03 program9 (Fig. S3, ESI†). The pattern of absorption spectrum of 1a computed from TDDFT calculations10 in aqueous solution, specially the newly generated band ca. 404 nm for HOMO (104A) → LUMO (105A) excitation nicely matches with the experimental spectrum (Fig. 2B). The existence of monomer (1a) was further supported by the mass analysis of 1 (ESI-MS+: m/z 521.37 for [1aH]+; 543.36 for [1aNa]+) (Fig. S4A, ESI†). Again, identical CT band ca. 400 nm for 1 and corresponding in situ complex, 1
with Z = 2 to exist as dimer in solid phase (Fig. 1) (ESI†). In aqueous medium, the dimeric core probably breaks up into two monomeric units. The most probable monomeric structure (1a) in aqueous solution was optimized in gas phase by DFT calculation using Gaussian 03 program9 (Fig. S3, ESI†). The pattern of absorption spectrum of 1a computed from TDDFT calculations10 in aqueous solution, specially the newly generated band ca. 404 nm for HOMO (104A) → LUMO (105A) excitation nicely matches with the experimental spectrum (Fig. 2B). The existence of monomer (1a) was further supported by the mass analysis of 1 (ESI-MS+: m/z 521.37 for [1aH]+; 543.36 for [1aNa]+) (Fig. S4A, ESI†). Again, identical CT band ca. 400 nm for 1 and corresponding in situ complex, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry in solution confirming from fluorescence quenching measurement and Job's method (Fig. S1 and S5, ESI†) indicate that 1 produces same species in aqueous solution which was generated during Cu(BP)(ClO4)2 vs. HHMB titration (vide supra); although unlike 1, small residual fluorescence during in situ generation at saturated condition was probably due to some unreacted HHMB (Fig. S1A, ESI†). Complete fluorescence quenching of 1 finally renders its applicability to act as the sensor preferable over its in situ generation.
:1 stoichiometry in solution confirming from fluorescence quenching measurement and Job's method (Fig. S1 and S5, ESI†) indicate that 1 produces same species in aqueous solution which was generated during Cu(BP)(ClO4)2 vs. HHMB titration (vide supra); although unlike 1, small residual fluorescence during in situ generation at saturated condition was probably due to some unreacted HHMB (Fig. S1A, ESI†). Complete fluorescence quenching of 1 finally renders its applicability to act as the sensor preferable over its in situ generation.
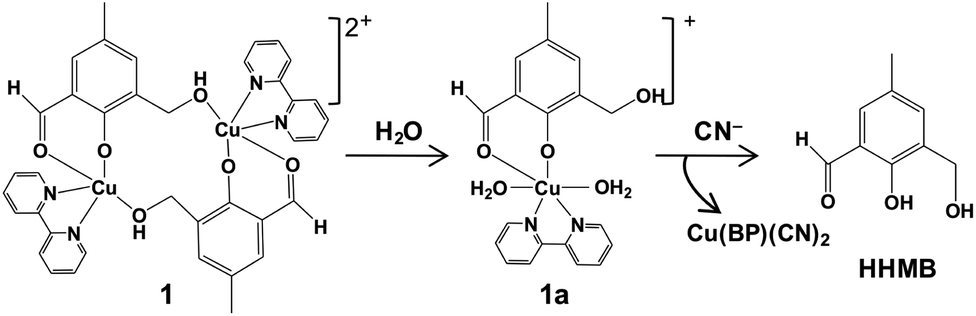 | ||
| Scheme 1 Cyanide-induced Cu(II) displacement mechanism for 1. | ||
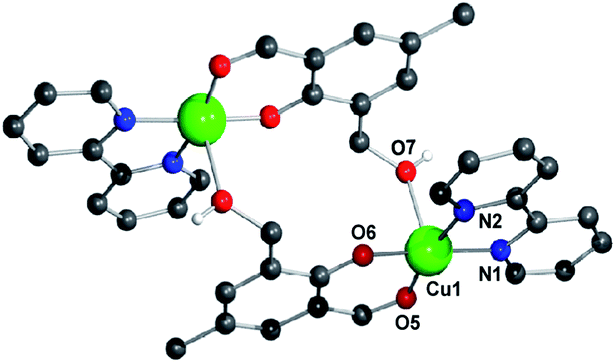 | ||
| Fig. 1 Molecular view of sensor 1 with atom numbering scheme. All H-atoms except that for O7 and ClO4− are omitted for clarity. Color index: C (black), N (blue), O (red), Cu (green). | ||
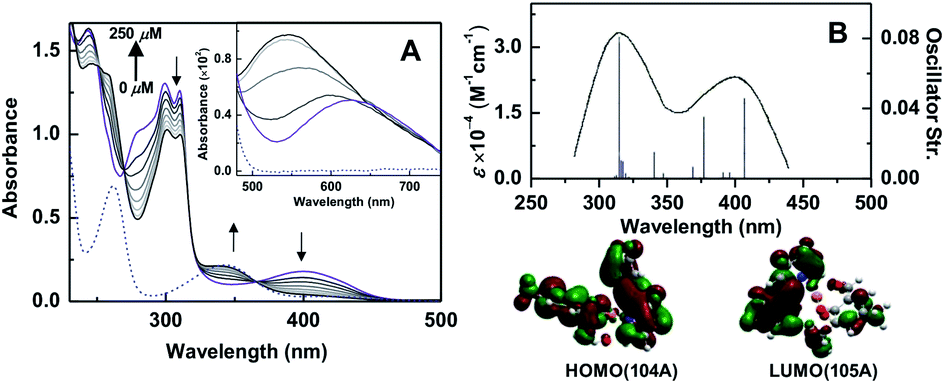 | ||
| Fig. 2 (A) UV-Vis absorption spectra of 1 (50 μM) in presence of increasing CN− concentration (0–250 μM) in 20 mM HEPES, pH 7.4. (Inset) spectra of 1 (100 μM) in presence of CN− (0–500 μM) are shown. The dashed blue line for HHMB (50 μM) is used as reference. CN− free spectrum of 1 is indicated by purple color. (B) TDDFT spectrum (250–450 nm) in water and FMO (below) for 404 nm CT transition of 1a. | ||
To determine the selective cyanide sensing ability, sensor 1 (20 μM, with respect to HMB unit) was used as completely non-fluorescent “turn-off” mode in 100% aqueous buffer at physiological pH 7.4 (Fig. 3A, green curve). Instantaneous CN−-induced fluorescence intensity was increased sharply ca. 10-fold at 10 μM CN− (inset, Fig. 3C). The increase of intensity continued until saturation was obtained ca. 100 μM of CN− (Fig. 3A) and we wonder that it took ca. 400 μM CN− to reach the saturation for in situ generated complex of 1 (Fig. 3B). For selectivity, similar experiments were performed under identical conditions with various potentially interfering anions (N3−, OAc−, HPO42− etc.) as well as bio-disturbing molecules (ATP, glutathione, cysteine, urea etc.), and failed to recover any noticeable fluorescence, however, 1 recognized selectively CN− from the mixture of various other anions/molecules with almost identical accuracy and hence CN− sensing ability of 1 is confirmed unambiguously. (Fig. 3C and S6, ESI†). Interestingly, identical fluorescence characteristic of free HHBM and that for 1 + CN− in “turn-on” mode, confirms the liberation of free fluorophoric HHMB by the CN−-induced decomplexation. Indeed, the identical excited state lifetimes (ca. 4.8 ns) for free HHBM and sensor 1 containing 5-equivalent CN− also support this proposition (Fig. S7, ESI†). The maximum increase of emission intensity ratio ca. 115-fold by CN− (Fig. 3C), was much higher than those for in situ complexes, viz. ca. 18 and 4 for Cu(BP)(ClO4)2 and Cu(ClO4)2 respectively (Fig. S8, ESI†). Unlike allied in situ complexes, excellent co-linearity for the change in emission intensity for 1 with CN− concentration up to 4-equivalent in 100% aqueous medium at physiological pH can be utilized as a good ratiometric chemosensor in biochemical systems (inset Fig. 3C and S8D, ESI†). The ratiometric value was almost remaining constant in the pH range 6.5–8.0 (Fig. S9, ESI†) as well as insensitive towards other common interfering metal ions (Na+, K+, Ca2+, Zn2+, Mg2+ etc.) present in biological systems. The obtained limit of detection (LOD) ca. 0.7 μM is well below the WHO permissible limit (ESI†) and hence 1 can be an excellent choice for even below μM CN− detection.
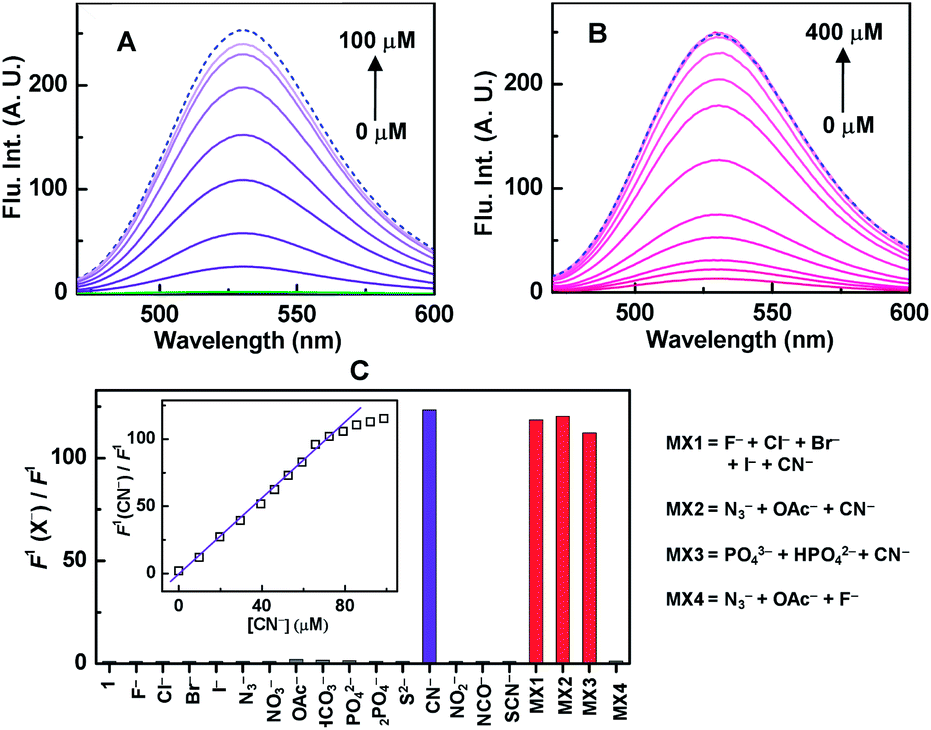 | ||
| Fig. 3 Fluorescence response of (A) 1 (20 μM), (B) HHMB (20 μM) + Cu(BP)(ClO4)2 (120 μM) towards increasing CN− concentration in 10 mM HEPES, pH 7.4. The green spectrum (A) is for 1 in absence of CN−. The dashed blue line for HHMB (20 μM) is used as reference. (C) Ratio of fluorescence intensity of 1 (20 μM) in presence (F1(CN−)) and absence (F1) of various anions (100 μM) or mixture of anions (100 μM for each anion) are depicted by bar-diagram. (Inset) the ratio for 1 at different CN− concentrations are plotted. (Excitation: 440 nm.) | ||
The chemical changes during CN−-induced decomplexation were also investigated by the UV-Vis absorption studies. A distinct reduction of the CT band ca. 400 nm for 1 was observed with increasing CN− concentration along with the concomitant formation of a new absorption band at a similar position ca. 340 nm of HHMB absorption (Fig. 2A). The CN−-induced decomplexation for 1 also accompanied by isobestic points at ca. 367, 316, 269 and 251 nm indicates the generation of free HHMB. To ascertain the Cu(II) coordination of 1 during displacement, we compared the changeover for CN−-induced d–d absorption profile of 1 and Cu(BP)(ClO4)2 respectively. In both cases, close resemblance of the d–d absorption spectra confirmed the identical Cu(II) coordination either in the displaced Cu(II) from 1 (inset Fig. 2A) or in Cu(BP)(ClO4)2 (Fig. S10, ESI†). Moreover, the absence of characteristic absorption spectrum of free BP for Cu(BP)(ClO4)2 in presence of CN− (Fig. S10, ESI†), assured the direct attachment of CN− to the Cu(II) centre in 1 to produce [Cu(BP)(CN)x]n−. In fact, the ESI mass analysis of 1 in presence of 5-equivelent CN− confirmed the generation of Cu(BP)(CN)2 (ESI-MS+: m/z 278.68 for [Cu(BP)(CN)2Li]+) (Fig. S4B, ESI†).
To demonstrate the bio-applicability of sensor 1 for CN− detection, the nematode C. elegans was used for in vivo imaging. It is the most suitable organism for testing the cyanide toxicity of municipal and industrial waste water.6a No fluorescence was observed in C. elegans when it was exposed to 1 (20 μM) (Fig. 4A). When the nematodes were exposed to 4 μM CN− in presence of 1 (20 μM), a trace amount of fluorescence was mainly observed in the peripheral region (Fig. 4B). However, when the concentration of CN− was increased to 100 μM under same condition, strong fluorescence was observed throughout the whole body of the nematode (Fig. 4C). Noticeably, compared to 1, such in vivo imaging for cyanide detection was relatively less sensitive when nematodes were exposed to allied in situ complex, followed by CN− addition (Fig. 4D–F and S11, ESI†). Furthermore, assessment of toxicity in the form of lethality assays11 with the wild type worms revealed no effect on the survival of the worms even in higher 1 mM doses of the sensor 1 as well as the HHMB (Table S4, ESI†). Thus our probe 1 deserves a novel status for in vivo imaging under physiological conditions.
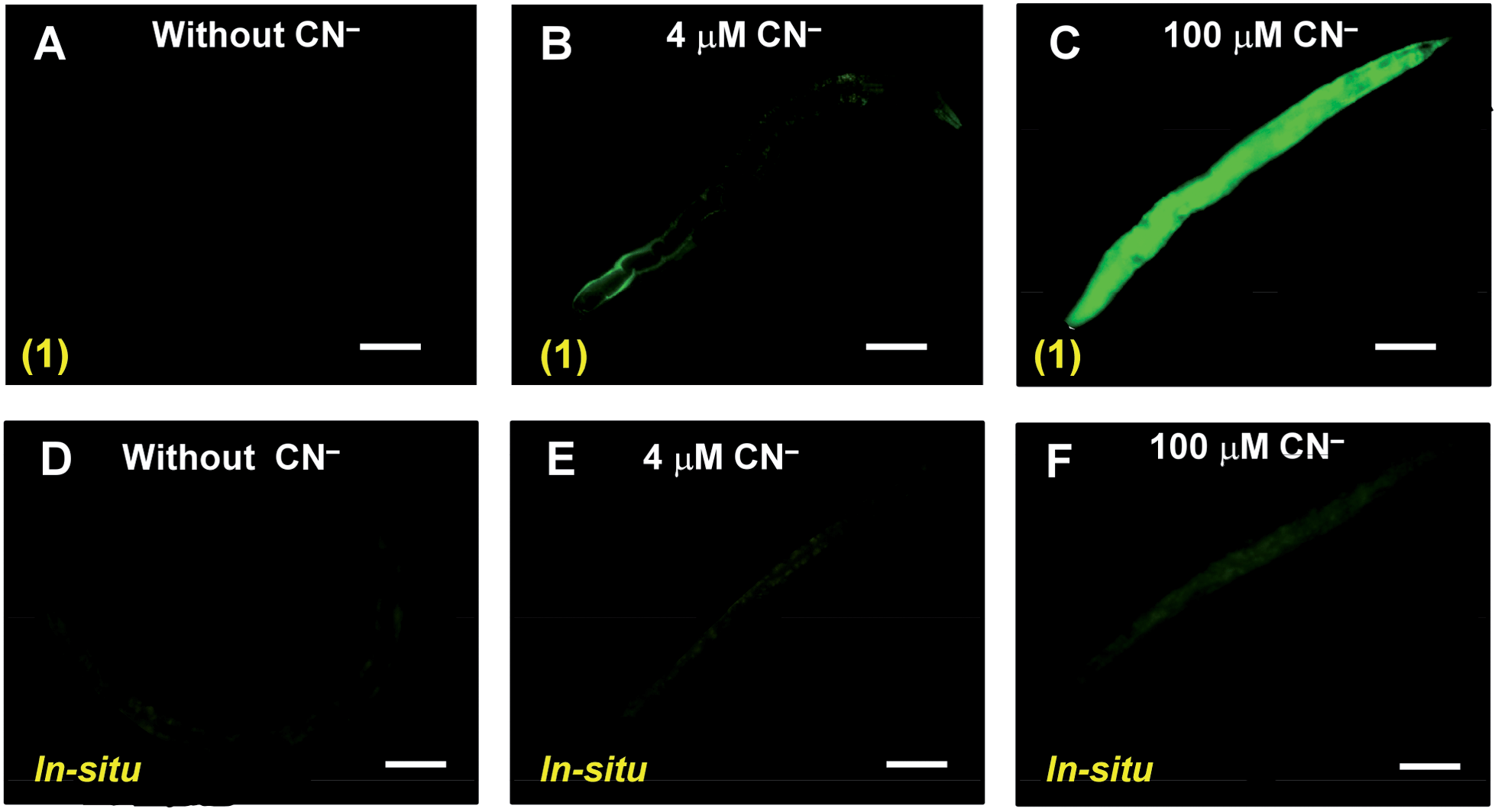 | ||
| Fig. 4 Fluorescence images of the nematode C. elegans exposed to complex 1 (A–C) and in situ complex between HHMB and Cu(BP)(ClO4)2 (D–F) in presence of different CN− concentration. The phase contrast is identical for all images. The scale bars: 40 μm. | ||
In conclusion, we have synthesized a nontoxic fluorescent chemosensor [Cu(BP)HMB]2(ClO4)2 (1), that remains in monomeric form (1a) in aqueous medium. The choice of Cu(BP)(ClO4)2 instead of Cu(ClO4)2 for in situ complex generation is justified. However, the superior selectivity, sensitivity and bio-applicability of 1 for sensing CN− in 100% water under physiological conditions compared to allied in situ complexes renders novelty and to the best of our knowledge, no such analytical comparison was reported so far. Below μM LOD and rapid ratiometric response of the sensor are also advantageous. For CN− sensing fluorescence “off–on” mechanism, we report the generation of [Cu(BP)(CN)x]n− species for the first time during the displacement approach.
Authors acknowledge JU and Maulana Azad College for departmental facilities, Dr A. Bandyopadhyay, IICB Kolkata for imaging facilities. JB thanks CSIR (Grant: 37-1486/11/EMR-II).
Notes and references
- (a) X. Lou, D. Ou, Q. Li and Z. Li, Chem. Commun., 2012, 48, 8462 RSC; (b) J. Du, M. Hu, J. Fan and X. Peng, Chem. Soc. Rev., 2012, 41, 4511 RSC; (c) P. A. Gale, N. Busschaert, C. J. E. Haynes, L. E. Karagiannidis and I. L. Kirby, Chem. Soc. Rev., 2014, 43, 205 RSC.
- (a) J. Taylor, N. Roney, C. Harper, M. Fransen and S. Swarts, Toxicological Profile for Cyanide, US Department of Health and Human Services, Atlanta, GA, 2006, pp. 6–7 Search PubMed.
- Guidelines for Drinking-Water Quality, ed. M. Sheffer, World Health Organization, Geneva, 1996 Search PubMed.
- (a) G. C. Miller and C. A. Pritsos, Cyanide: Social, Industrial and Economic Aspects, Proceedings of the TMS Annual Meeting, 2001, pp. 73–81 Search PubMed.
- (a) Z. Xu, X. Chen, H. N. Kim and J. Yoon, Chem. Soc. Rev., 2010, 39, 127 RSC; (b) B. B. Shi, P. Zhang, T. B. Wei, H. Yao, Q. Lin and Y. M. Zhang, Chem. Commun., 2013, 49, 7882 Search PubMed.
- (a) S.-Y. Chung, S.-W. Nam, J. Lim, S. Park and J. Yoon, Chem. Commun., 2009, 2866 RSC; (b) H. S. Jung, J. H. Han, Z. W. Kim, C. Kang and J. S. Kim, Org. Lett., 2011, 13, 5056 CrossRef CAS PubMed; (c) M. Wang, J. Xu, X. Liu and H. Wang, New J. Chem., 2013, 37, 3869 RSC.
- (a) Z. Yang, Z. Liu, Y. Chen, X. Wang, W. He and Y. Lu, Org. Biomol. Chem., 2012, 10, 5073 RSC; (b) S. Madhu, S. K. Basu, S. Jadhav and M. Ravikanth, Analyst, 2013, 138, 299 RSC; (c) S. Goswami, A. Manna, S. Paul, A. K. Das, K. Aich and P. K. Nandi, Chem. Commun., 2013, 49, 2912 RSC.
- E. Lambert, B. Chabut, S. C. Noblat, A. Deronzier, G. Chottard, A. Bousseksou, J.-P. Tuchagues, J. Laugier, M. Bardet and J.-M. Latour, J. Am. Chem. Soc., 1997, 119, 9424 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, revision C. 02, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- M. Dengg and J. C. A. van Meel, J. Pharmacol. Toxicol. Methods, 2004, 50, 209 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterizations. CCDC 960526. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra00069b |
| This journal is © The Royal Society of Chemistry 2014 |