Quantitative detection of trace mercury in environmental media using a three-dimensional electrochemical sensor with an anionic intercalator†
Yi Zhangab,
Guang-Ming Zeng*ab,
Lin Tang*ab,
Yuan-Ping Liab,
Zhao-Meng Chenab and
Guo-He Huangab
aCollege of Environmental Science and Engineering, Hunan University, Changsha 410082, P.R.China. E-mail: zgming@hnu.edu.cn; tanglin@hnu.edu.cn; zyi@hnu.edu.cn; Fax: +86-731-88823701; Tel: +86-731-88822754
bKey Laboratory of Environmental Biology and Pollution Control (Hunan University), Ministry of Education, Changsha 410082, P.R.China
First published on 28th February 2014
Abstract
Mercury, one of the most widespread highly toxic heavy metals, has severe detrimental effects on human health and the environment. It is significant to develop a sensitive and reliable method of accurately detecting trace levels of mercuric ions in environmental media to meet the ever-increasing demands of ongoing environmental monitoring programs. A three-dimensional mercuric ion sensor was constructed using mercury-specific oligonucleotides, gold nanoclusters, and an anionic intercalator. Due to the steric reaction field in the electrode surface microenvironment formed by the gold nanoclusters, the sensor could spatially capture mercuric ions with electroactive indication to realize trace mercury measurements with high sensitivity; the sensor exhibited strong environmental adaptability, high selectivity, and other advantages. Under optimal conditions, mercuric ions could be detected in the range from 0.05 to 350 nM, and the detection limit was 0.01 nM. The mercuric ion sensor was compared with atomic fluorescence spectrometry to analyze municipal wastewater and river water samples. In addition, the sensor performance was also analyzed according to derived formulae. This accurate method has the potential to be deployed in the field for measurements of mercury in environmental media.
Introduction
Heavy metal pollution is a global environmental issue due to its severe effect on human health and the environment.1–3 As the notorious culprit behind Minamata disease, mercury has become a focus within the family of heavy metals.1,2 The Governing Council of the United Nations Environment Programme agreed on the need to develop a global legally binding instrument on mercury in 2009, and the work to prepare the instrument was undertaken by the intergovernmental negotiating committee.4 Mercury pollution comes mainly from coal combustion, industrial production and agricultural chemicals. Annual total global mercury emission from nature and human activities is approximately 7500 tons per year.5 Mercury exists in the environment in a variety of forms, for instance, as the water-soluble mercuric ion (Hg2+). Mercuric ions are an important form with a high toxicity in the mercury system. Moreover, mercuric ions in aquatic sediments can be converted into the more toxic methyl mercury, which can accumulate in the human body through the food chain and lead to permanent damage to the brain, and to other chronic diseases.1,6,7 In 2005, the U.S. Environmental Protection Agency set the maximum allowable level for mercuric ions in drinkable water as 10 nM.8 Therefore, monitoring mercury is of critical importance in environmental and food safety, as well as clinical toxicology.The methodology of mercury detection has undergone an unceasing development. Traditionally, mercury is detected by using inductively coupled plasma mass spectrometry (ICP-MS),9 atomic absorption–emission spectroscopy,10 and cold vapor atomic fluorescence spectrometry.11 However, sophisticated instrumentation and/or complicated sample preparation processes limit their application to on-site mercury control. Currently, the mercury-specific oligonucleotide technique for identification and quantification of mercury has attracted great attention.12–21 The mercury-specific oligonucleotide technique is based on thymine–Hg2+–thymine (T–Hg2+–T) coordination chemistry, and the resulting Hg2+-stabilized hybridization of oligonucleotides with T–T mismatches has a specific recognition ability of Hg2+ with convincing detection accuracy.12 The combination of an electrochemical sensor and a mercury-specific oligonucleotide is a competitive strategy. Electrochemical sensors have become a research focus since some advantages e.g. inherent simplicity, low cost, high sensitivity and selectivity etc., were observed.22 In particular, their excellent compatibility with miniaturization technology leads to future tendencies of development.22–24 Indeed, since Ono and Togashi12 observed the specific binding of Hg2+–thymine complexes in DNA and developed a sensor based on fluorescence resonance energy transfer with a detection limit of 40 nM, various mercury detection strategies based on this property have been developed in recent years. Among them, electrochemical sensors based on electrical activity indicators or electrically contacted enzymes, are relatively convenient and stable, and have showed significant plasticity and high sensitivity.17–19
Nanomaterials can improve sensor performances significantly.25,26 Among many nanomaterials, nano-Au frequently appears in sensor configurations, especially in DNA sensors, due to its strong electron transfer capacity, high biocompatibility, and stable thiol–Au affinity.27,28 Gold nanoclusters have recently been developed from gold nanoparticles, which not only keep the inherent properties of nano-Au, but also exhibit spatiality when they are used to modify a supporter surface, forming spinous structures with certain lengths. Previously this spatiality was observed by atomic force microscopy.29 Notably, the configuration of most sensors, including those based on nano-Au, is generally a two dimensional planar structure, and the configuration of those based on gold nanoclusters is three dimensional (3D). It is believed that a sensor with a 3D configuration can obtain a more sensitive signal response due to its larger specific surface area and increased number of reaction sites. Though gold nanoclusters have been used in immunosensors,30 there are few reports about their application in DNA sensors, and little attention has been paid to mercuric ion sensors based on mercury-specific oligonucleotides.
In addition, the choice of signal agent is also important for the construction of an electrochemical sensor. A proper signal agent is as of much benefit to the whole working of the sensor and a stable signal response as the original design. For a DNA sensor, it is believed that an electroactive intercalator, as a label-free indicator, is an ideal candidate because it not only provides enough electronic response, but also contributes to a more flexible, simple, and stable sensor strategy. From the viewpoint of the control of false positive responses and background signal, anionic intercalators show better effects than cationic intercalators in DNA analysis.31 Therefore, an anionic intercalator was used for the proposed electrochemical sensor.
Herein, mercury-specific oligonucleotides, three dimensional gold nanoclusters, and an anionic intercalator were used to construct a highly sensitive sensor for mercury detection in environmental samples, which has not been reported so far to the best of our knowledge. This strategy for mercury detection is relatively simple, operates with high accuracy, and exploits strong environmental impact resistance. The gold nanoclusters were electrodeposited on a gold electrode surface forming a 3D structure, and then modified with single-stranded mercury-specific oligonucleotide probes (P1). In the presence of Hg2+, the P1 forms a double-stranded hairpin structure due to T–Hg2+–T mismatches. Meanwhile, the anionic intercalator, disodium-anthraquinone-2,6-disulfonate (AQDS), was selected as an electroactive signal indicator because of its low background noise and good electrochemical performance. Furthermore, mercury detection in environmental samples was performed to investigate and demonstrate the application of the proposed sensor.
Experimental
Apparatus
Electrochemical measurements were carried out on a CHI660B electrochemistry system (Chenhua Instrument, Shanghai, China). The three-electrode system used in this work consists of a gold electrode (3 mm in diameter) as the working electrode, a saturated calomel electrode (SCE) as a reference electrode and a Pt foil auxiliary electrode. A track-etched polycarbonate (0.2 μm) membrane was provided by Whatman (UK). Scanning electron micrographs (SEM) of the morphology of the electrode surface were obtained with a JSM-6700F field emission scanning electron microscope (JEOL Ltd., Japan). A Sigma 4K15 laboratory centrifuge, a Sigma 1-14 microcentrifuge (Sigma, Germany), an AFS-9700 atomic fluorescence spectrophotometer (Kechuang Haiguang Instrument, Beijing, China) and a model CS501-SP thermostat (Huida Instrument, Chongqing, China) were used in the assay. All work was performed at room temperature (25 °C) unless otherwise mentioned.Reagents
Disodium-anthraquinone-2,6-disulfonate (AQDS) was purchased from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan). Hydrochloroauric acid was supplied by Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Ascorbic acid (AA), 6-mercapto-1-hexanol (MCH) and mercury nitrate were purchased from Sigma-Aldrich Chemical Co. All chemicals were of analytical grade. This work used a Tris–HCl buffer (10 mM Tris adjusted to pH 8.0 with 10 mM HCl) and phosphate buffered saline (PBS, 0.07 M KH2PO4 and 0.07 M Na2HPO4). All solutions were prepared in deionized water of 18 MΩ purified using a Milli-Q purification system.The synthesized oligonucleotides, all HPLC-purified and lyophilized, were provided by Sangon Biotech. Co., Ltd. (Shanghai, China). The sequences were as follows:
5′-SH-(CH2)6-TCA TGT TTG TTT GTT GGC CCC CCT TCT TTC TTA-3′ (P1)
Probe 1 (P1) was dissolved in Tris–HCl buffer containing 1 M NaCl and kept at −20 °C for further use. The 1 mM AQDS was stored in PBS (pH 7.0) containing 0.2 M NaCl, in the dark.
Sensor fabrication
The gold working electrodes were polished thoroughly with 300 nm and 50 nm alumina slurry and sonicated in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) HNO3, acetone, and water successively to remove the residues. Then the gold electrodes were electrochemically cleaned in 0.5 M H2SO4 by cyclic voltammetry between −0.4 and 1.5 V (vs. SCE) until a steady state redox wave was observed. Afterwards, Au nanoprickle clusters were electrodeposited onto the gold electrode.31 The polycarbonate membrane was carefully attached onto the electrode surface and fixed by a rubber O-ring. Electrodeposition was performed using chronoamperometry in 1% (w/w) HAuCl4 solution containing 0.5 M perchloric acid at a potential of 0.18 V for 120 s. Following immersion in chloroform to dissolve the polycarbonate template, the obtained electrode was thoroughly rinsed with water. Next, an 8 μL aliquot of the thiolated probe (P1, 4 μM) was pipetted onto the electrode surface and underwent self-assembly through thiol–gold bonding for 3 hours. After adsorption, the electrode was copiously rinsed with Tris–HCl buffer containing 0.5 M NaCl and water to remove any non-specific adsorbed materials. Following adsorption of a MCH monolayer in 1 mM ethanolic solution for 30 min, the prepared electrode was thoroughly rinsed with Tris–HCl buffer containing 0.5 M NaCl. When not in use, the electrode was stored in a moist state at 4 °C.
:1 (v/v) HNO3, acetone, and water successively to remove the residues. Then the gold electrodes were electrochemically cleaned in 0.5 M H2SO4 by cyclic voltammetry between −0.4 and 1.5 V (vs. SCE) until a steady state redox wave was observed. Afterwards, Au nanoprickle clusters were electrodeposited onto the gold electrode.31 The polycarbonate membrane was carefully attached onto the electrode surface and fixed by a rubber O-ring. Electrodeposition was performed using chronoamperometry in 1% (w/w) HAuCl4 solution containing 0.5 M perchloric acid at a potential of 0.18 V for 120 s. Following immersion in chloroform to dissolve the polycarbonate template, the obtained electrode was thoroughly rinsed with water. Next, an 8 μL aliquot of the thiolated probe (P1, 4 μM) was pipetted onto the electrode surface and underwent self-assembly through thiol–gold bonding for 3 hours. After adsorption, the electrode was copiously rinsed with Tris–HCl buffer containing 0.5 M NaCl and water to remove any non-specific adsorbed materials. Following adsorption of a MCH monolayer in 1 mM ethanolic solution for 30 min, the prepared electrode was thoroughly rinsed with Tris–HCl buffer containing 0.5 M NaCl. When not in use, the electrode was stored in a moist state at 4 °C.
Detection strategy
A succinct strategy was designed to detect Hg2+ ions using the prepared electrode (see Fig. 1). The electrode was directly reacted with a Hg2+ sample for 60 minutes to form T–T mismatched dsDNA on the electrode surface and was then immersed into AQDS solution for electrochemical detection. AQDS has a unique anthracene ring structure, can intercalate into the duplex structure of dsDNA, and exhibits a reversible 2-electron transfer process for its quinone/hydroquinone redox couple in electrochemistry.31 | ||
| Fig. 1 Schematic diagram of the detection strategies for mercuric ion detection. | ||
Electrochemical detection
To investigate the performance of the Hg2+ sensor, cyclic voltammetry (CV) and square wave voltammetry (SWV) were executed in 20 mL of PBS containing 0.3 M NaCl. The CV was performed between −0.4 and 0.4 V vs. SCE at 100 mV s−1, and the SWV was carried out between −0.2 and −0.7 V under a pulse amplitude of 25 mV and a frequency of 10 Hz, with a step potential of 4 mV. The corresponding peak current was recorded.Analysis of environmental samples
Two kinds of typical environmental samples, municipal wastewater and river water samples, were used in this study. The municipal wastewater was obtained from a municipal wastewater treatment plant in Changsha, China. The river water was from the Xiangjiang river in Changsha, China. They were centrifuged at 10000 rpm for 5 minutes and filtered to remove any suspension and solid impurities. Following spiking with different concentrations of mercuric ions, the resulting samples were analyzed by the supposed sensor. Meanwhile, the same samples were filtrated via a 0.2 μm polycarbonate filter and analyzed by atomic fluorescence spectrometry (AFS) for comparison.
Results & discussion
Characteristics of the electrode
The white polycarbonate template turned yellow after 120 s of electro-deposition, and concomitant with the dissolving of the template, the surface morphology of the electrode changed from mirror-like to barbed-tongue-like by naked-eye observation, indicating that the successful electrodeposition of Au nanoprickle clusters took place. Further characterization of the electrode surface morphology was carried out by SEM. As shown in Fig. 2, significant variations in the surface morphology can be observed before and after the electrode modification. Compared with the relatively smooth surface of the original electrode, that of the modified electrode becomes granular and uneven. The Au nanoprickles were electrodeposited onto the electrode surface, and protuberant clusters (bright points) grew along the pores of the polycarbonate template. The mean diameter of the Au nanoclusters was around 120 nm. | ||
| Fig. 2 SEM images of (a, b) the original electrode surface, and (c, d) the Au nanoprickle modified electrode surface. | ||
With the help of thiol–gold bonding and the effect of MCH, the thiolated probes stretch over the gold medium like tentacles. Unlike the 2D planar structure of a conventional electrode surface, the Au nanoprickle clusters developed a 3D spatial structure in the electrode surface microenvironment, and formed a steric reaction field with an increase in reaction space, which is beneficial for the shuttling of Hg2+ ions and indicating capture by the DNA probes. This characteristic theoretically improves the detecting sensitivity.
The electrochemical response capacity of the electrodes was investigated via electrochemical methods. Under the same experimental conditions and processes, the peak current responses from square wave voltammetry are compared in Fig. 3. It was found that the peak current response increased significantly from the original electrode to the modified electrode. In view of this, the modified electrode is superior to the conventional electrode due to the effect of the 3D nanoclusters.
 | ||
| Fig. 3 Square wave voltammograms of the modified electrode and the original gold electrode between −0.2 and −0.7 V under a pulse amplitude of 25 mV and a frequency of 10 Hz with a step potential of 4 mV in 20 mL of PBS containing 0.3 M NaCl (pH 7.0), after reacting with 100 nM Hg2+ ion for 60 minutes and immersing into AQDS solution containing 0.2 M NaCl for 360 minutes. | ||
Detectivity
The effect of the 3D nanoclusters can be further analyzed according to the notion of detectivity. The detectivity ε of a sensor is defined as the quotient of the normalized signal output Y over the normalized signal input X, i.e.32
 | (1) |
| ΔY/ΔX = be−bX/N | (2) |
| b = e−U/kT | (3) |
| σX2 = xN = X | (4) |
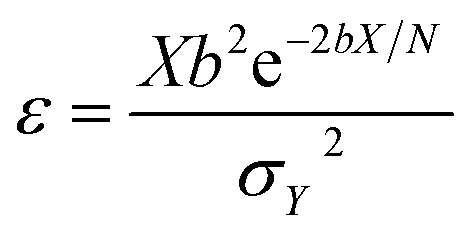 | (5) |
Therefore, increasing the surface area and the number of reaction sites, i.e. the number of receptors covering the sensor surface N, can raise the detectivity.
The gold nanoclusters were electrodeposited onto the proposed sensor surface. Due to forming a 3D structure on the electrode surface, a larger surface area and more reaction sites were obtained. The total surface area was estimated to be 5.0 times larger than that of the original electrode surface (see the ESI†). Hence the detection performance was improved. This result can also explain the aforementioned electrochemical behaviors.
Choice of electroactive indicator
Electroactive indicators are frequently used in the detection of hybridization or base mismatch of synthesized short-stranded DNA, because they can intercalate into the duplex structure of dsDNA but not into ssDNA, and express electrical activity. The indicators are classified into cationic and anionic intercalators. It should be noted that both dsDNA and ssDNA carry negative charges and will attract the cationic intercalators due to electrostatic attraction. Indeed, the interaction between the cationic intercalators and dsDNA is a synergistic action of electrostatic attraction, intercalation, and perhaps physical adsorption. Therefore, a big background signal is unavoidable. In particular, when the electrostatic attraction becomes the predominant force in the interaction, it is difficult to distinguish between dsDNA and ssDNA. In fact, although cationic intercalators, e.g. methylene blue, cobalt metal complexes [Co(bpy)3]3+, rhodium metal complexes [Rh(phi)dmb]3+, ruthenium metal complexes [Ru(phen)2(dppz)]2+, and others,33 have been applied more commonly, the exhibition of large background signal and relatively poor accuracy always prevented sensitivity enhancement in DNA analysis. Alternatively, anionic intercalators can eliminate false positive responses and reduce background signal, because the effect of electrostatic attraction between the two types of DNA and anionic intercalators does not exist. Gooding and co-workers31 proved that anionic intercalators, e.g. AQDS, behaved better than cationic ones. AQDS has a perfectly symmetric anthraquinone ring structure, and exhibits a reversible 2-electron transfer process for its quinone/hydroquinone redox couple in electrochemistry. Inspired by this property, AQDS was chosen as the electroactive indicator for the duplex structure of dsDNA obtained from T–Hg2+–T mismatch.Optimization of the detection strategy
A series of experiments was performed to optimize the conditions to give acceptable signal response. The effects of pH value, the amount of single stranded probe P1, the self-assembly time, the reacting time with the Hg2+ ions, the intercalation time of the AQDS and the ionic strength of the AQDS solution were investigated. The effect of temperature was not considered, because it is our intention that the Hg2+ sensor will be used at room temperature, which is more suitable for application.pH always plays an important role in chemical reactions, especially in biochemical reactions. The working principle of the sensor involved the bioactivity of the DNA probe, the process of T–Hg2+–T mismatch, and the AQDS intercalation, which were affected by the pH. Considering that the pH conditions of some links, e.g. the self-assembly of the DNA probe, the AQDS intercalation, and the electrochemical detection, were fully controllable, and only that of the real Hg2+ ion sample had a certain unpredictability, samples with different pH values containing 10 nM of Hg2+ ions were used to investigate the pH effect. Although acidic conditions were beneficial to the freeness of the Hg2+ ions, the DNA would be damaged once a certain limit was exceeded. Under alkaline conditions, the bioactivity of the DNA is maintained well in the appropriate range, but the freeness of the Hg2+ ions reduces. It can be seen in Fig. 1S† that the current responses were not only similar but also the maximum current responses appeared in the pH range 6.0–7.8. This was a gratifying phenomenon that revealed that the sensor had a good ability to resist the impact of pH change. Furthermore, the pHs of river water (pH = 6.0–6.9) and municipal wastewater (pH = 6.5–7.5) are generally within this range. The above indicated that the sensor was quite suitable for real sample detection. In order to ensure experimental consistency, the pH of the related solutions is 7.0 unless otherwise mentioned.
With regard to the probes immobilizing on the surface of the electrode, the ideal state is that they simultaneously occupy the surface contact points as much as possible, and are evenly distributed with enough space among them to maximize the capture of the target, improving the sensitivity of the sensor. Varying concentrations of P1 (1–6 μM) were used to self-assemble for 60, 90, 120, 150, 180, 210, and 240 minutes and compared to find the desired result. As shown in Fig. 2S,† the most efficient result was obtained when 4 μM of P1 self-assembled in 180 minutes.
The reaction time between P1 and the Hg2+ ions can directly affect the detecting result and the efficiency. Theoretically, if the reaction is inadequate, the obtained product is not enough to achieve the best result; but if the reaction is too extensive, much time is wasted, reducing the detection efficiency. After testing, 60 minutes was chosen as the reacting time between P1 and the Hg2+ ions for the next experiments (see Fig. 3S†).
When AQDS is intercalated into the duplex structure of dsDNA, there is a certain resistance from electrostatic repulsion because both DNA and AQDS are negatively charged. Therefore, the intercalation process of AQDS is relatively slow. But suitably high salt concentration can speed up the intercalation by screening the electrostatic repulsion between the two. When the sensor is immersed in AQDS solution containing 0.2 M of NaCl for 360 minutes, the current response reached a maximum (see Fig. 4S†). However, from an efficiency point of view, an intercalation time of 240 minutes with 89% of the maximum current response was the optimal condition. Even so, the intercalation time was still longer than that needed for cationic intercalators. To meet the application requirements, therefore, a method of speeding up the intercalation of the anionic intercalators needs further study.
Response of the sensor to Hg2+ concentration
Under optimal detection conditions, Hg2+ would react with the T–T of P1 to form the duplex structure of dsDNA, into which AQDS would intercalate. As shown in Fig. 4, the peak current response attributed to the T–Hg2+–T mismatches increased with increasing Hg2+ concentration. The peak current response was linearly related to the logarithm of Hg2+ concentration C (nM) in the range from 0.05 to 350 nM by the following regression equation|
Current = (0.1163 ± 0.0038) × logC + (1.6270 ± 0.0062)
| (6) |
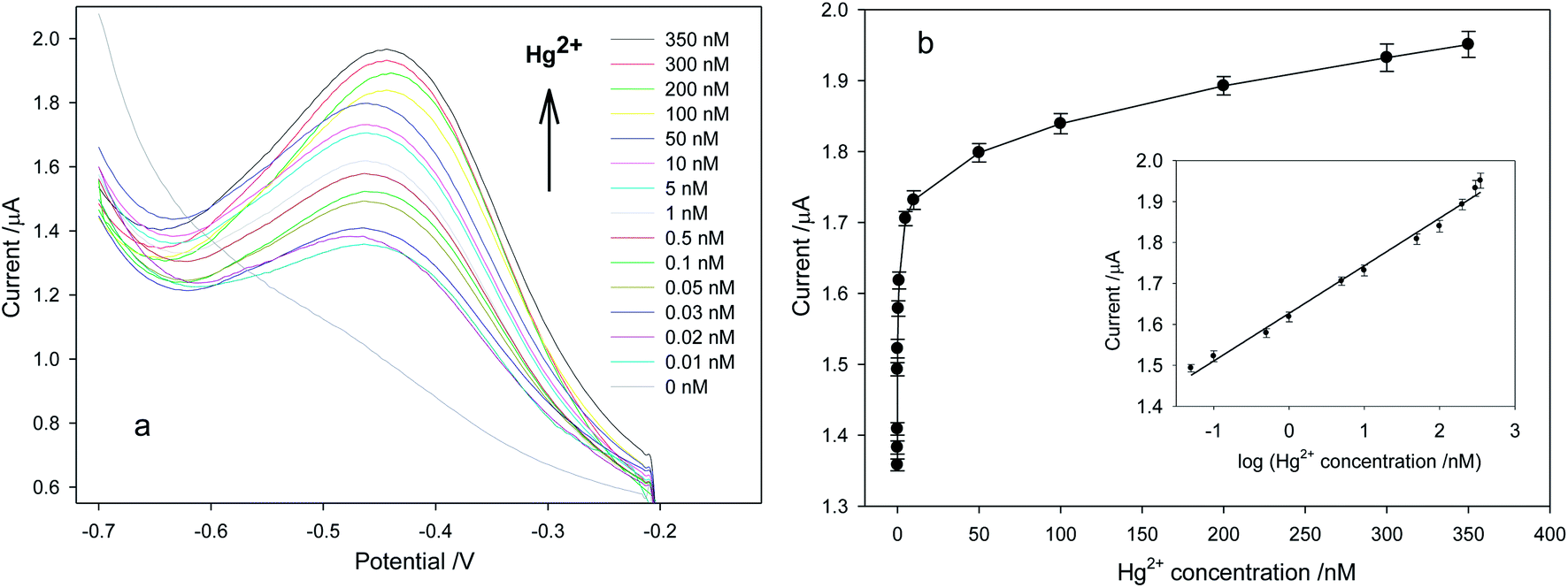 | ||
| Fig. 4 (a) Square wave voltammograms of the mercuric ion sensor for various concentrations of Hg2+ ions in 20 mL of PBS containing 0.3 M NaCl (pH 7.0) between −0.2 and −0.7 V under a pulse amplitude of 25 mV and a frequency of 10 Hz with a step potential of 4 mV. (b) The relative response current vs. Hg2+ ion concentration between 0.01 and 350 nM. The vertical bars designate the standard deviation from the mean of three replicate tests. Inset: calibration plot of the response current vs. the logarithm of Hg2+ ion concentration between 0.05 and 350 nM. | ||
The correlation coefficient was 0.9952. Each point of the calibration was done in triplicate, and the average relative standard deviation was 4.27%, which guaranteed the precision of the proposed sensor. The detection limit, according to the mean value of the background signal plus three times the standard deviation of the background signal, was calculated to be 0.00055 nM (n = 3; the number of blank runs). However, considering the actual detection results, the detection limit of Hg2+ ions was 0.01 nM. The proposed sensor had better sensitivity than previous electrochemical sensors (Table 1).17–19,34–36 It is apparent that the sensor was competent as an analysis tool for mercury detection in environmental media.
| Mercuric sensor | Range of detection (nmol L−1) | Limit of detection (nmol L−1) |
|---|---|---|
| Anodic stripping voltammetry of a switch conformation sensor17 | 1.0–250 | 0.5 |
| Cyclic voltammetry of a nanoengineered electrically contacted enzyme based sensor18 | 0.1–1000 | 0.1 |
| Square wave voltammetry of a supersandwich DNA based sensor19 | 0.1–1000 | 0.1 |
| Differential pulse voltammetry of a target-induced structure-switching DNA based sensor34 | 0.1–5000 | 0.06 |
| Differential pulse voltammetry of a Au NP functionalized DNA based sensor35 | 0.5–2000 | 0.5 |
| Square wave voltammetry of a Au NP amplified DNA based sensor36 | 0.5–100 | 0.5 |
| Present study | 0.01–350 | 0.01 |
Specificity and stability
A qualified detection method for environmental samples, having resistance to interference from irrelevant co-contaminants, is of importance and practical significance. Some environmentally relevant metal ions, including K+, Ba2+, Ca2+, Cd2+, Co2+, Cr2+, Cu2+, Mg2+, Mn2+, Ni2+, Pb2+, Zn2+, Al3+, Fe3+, and a mixture of these, were used to evaluate the performance of the mercuric ion sensor (see Fig. 5). It is shown that the interference of these environmentally relevant metal ions to the sensor is negligible. | ||
| Fig. 5 Interference test results using square wave voltammetry of the mercuric ion sensor in 20 mL of PBS containing 0.3 M NaCl (pH 7.0). Under optimal experimental conditions, 10 nM of Hg2+ with 500 nM of K+, Ba2+, Ca2+, Cd2+, Co2+, Cr2+, Cu2+, Mg2+, Mn2+, Ni2+, Pb2+, Zn2+, Al3+, Fe3+, and a mixture containing 10 nM of Hg2+ were tested. The vertical bars designate the standard deviation from the mean of three replicate tests. Table 1 gives a comparison of the sensitivity of different electrochemical mercuric ion sensors based on mercury-specific oligonucleotides. | ||
Good renewable capacity of the working electrode is beneficial to the performance stability of the sensor. As a regenerative agent, it is assumed that AA34 can weaken the coordination with T because it reduces Hg2+ to Hg+, and also EDTA37 can chelate Hg2+. Using only 100 nM of AA solution or 0.5 M of EDTA solution (pH = 9.0) to wash the working electrode for 60 minutes, the obtained regeneration efficiencies were only 57% and 65%, respectively. However, following treatment with EDTA solution for 40 minutes, and washing the electrode in AA solution for another 40 minutes, a regeneration efficiency of 93% was obtained. This effect obviously decreased after repeating the EDTA–AA regeneration 2–3 times. At this point, the electrode was treated in Piranha solution (a mixture of H2SO4 and H2O2 with a volume ratio of 3:2) at 60 °C for 30 minutes. The treated electrode reaches its original state and it is easy to modify the surface with the DNA probes once again. These regeneration operations can maintain the sensor stability and are suitable for practical applications.
Analysis of Hg2+ ions in samples
Municipal wastewater and river water samples spiked with Hg2+ ions were analyzed by the mercuric ion sensor. The pH of the municipal wastewater and the river water samples were 7.16 and 6.83, respectively, which were in the optimal pH range of the sensor (6.0–7.8). Therefore, the sensor was directly used to analyze the water samples after simple centrifugation and filtration operations. Simultaneously, the same samples, after being filtrated via a 0.2 μm polycarbonate filter and addition of a corresponding proportion of hydrochloric acid, were analyzed by AFS for comparison. The detection of Hg2+ ions from the samples was satisfactory and more stable results were obtained by the mercuric ion sensor, as presented in Table 2. Although the total time of the proposed method was longer than AFS, the actual machine detection time of the two methods was similar. Furthermore, compared with the limitations of the AFS method in application, e.g. tedious pretreatments, greater consumption of reagents, and a large instrument, the current method possessed certain advantages. The results indicated the potential of the sensor as a simple and reliable analysis method of Hg2+ ions in environmental samples.| Samples | Mercury concentrationa (nmol L−1) | Relative concentration deviation (%) | |
|---|---|---|---|
| Mercuric ion sensor | AFS | ||
| a An average of three replicate measurements ± standard deviation. | |||
| Municipal wastewater | 64.632 ± 0.258 | 65.539 ± 2.342 | 1.384 |
| 3.073 ± 0.036 | 3.056 ± 0.237 | 0.556 | |
| 5.309 ± 0.041 | 5.212 ± 0.059 | 1.861 | |
| River water | 7.428 ± 0.104 | 7.468 ± 0.073 | 0.536 |
| 134.872 ± 0.322 | 135.401 ± 0.419 | 0.391 | |
| 20.663 ± 0.067 | 20.412 ± 0.848 | 1.230 | |
Conclusion
In conclusion, a biosensor consisting of mercury-specific DNA, 3D gold nanoclusters and an anionic intercalator was developed, which provided the potential to quantify trace levels of mercuric ions in environmental water samples. 3D gold nanoclusters and the anionic intercalator significantly improved the detection performance of the sensor and it exhibited satisfactory results for Hg2+ ion detection in real environmental water samples. This method has strong environmental adaptability, high sensitivity, selectivity, appropriate renewable capacity and other advantages which meet the modern demand for mercury detection. We believe that the spatiality of the reaction field in the electrode surface microenvironment could be used for developing novel sensors. However, methods to speed up the intercalation of the anionic intercalator into the duplex structure of dsDNA need further investigation to improve the detection efficiency.Acknowledgements
This study was financially supported by the National Natural Science Foundation of China (nos 51039001, 50978088, 50908081, 51222805, 51278176), the fundamental Research Funds for the Central Universities, Hunan University, the Top Young Talents Program from Chinese Government, the Program for New Century Excellent Talents in University from the Ministry of Education of China (NCET-11-0129), the Hunan Key Scientific Research Project (2009FJ1010), and the Hunan Provincial Innovation Foundation For Postgraduate (CX2009B080).References
- H. H. Harris, I. J. Pickering and G. N. George, Science, 2003, 301, 1203 CrossRef CAS PubMed.
- A. H. Stern, Environ. Res., 2005, 98(1), 133–142 CrossRef CAS PubMed.
- N. Zhang, Q. Wang, X. Zhang, D. Zheng, Z. Zhang and S. Zhang, Sci. Total Environ., 2007, 387(1–3), 96–104 CrossRef PubMed.
- United Nations Environmental Program website, http://www.unep.org/hazardoussubstances/Mercury/Negotiations/INC2/tabid/3468/language/en-US/Default.aspx.
- Global mercury assessment, United Nations Environment Programme Chemicals, Geneva, Switzerland, 2002 Search PubMed.
- P. B. Tchounwou, W. K. Ayensu, N. Ninashvili and D. Sutton, Environ. Toxicol., 2003, 18(3), 149–175 CrossRef CAS PubMed.
- I. Onyido, A. R. Norris and E. Buncel, Chem. Rev., 2004, 104(12), 5911–5929 CrossRef CAS PubMed.
- Regulatory impact analysis of the final clean air mercury rule, EPA-452/R-05–003, U.S. Environmental Protection Agency, North Carolina, 2005.
- M. Wang, W. Feng, J. Shi, F. Zhang, B. Wang, M. Zhu, B. Li, Y. Zhao and Z. Chai, Talanta, 2007, 71(5), 2034–2039 CrossRef CAS PubMed.
- O. T. Butler, J. M. Cook, C. F. Harrington, S. J. Hill, J. Rieuwerts and D. L. J. Miles, J. Anal. At. Spectrom., 2006, 21(2), 217–243 RSC.
- Advances in atomic spectroscopy, ed. J. Sneddon, JAI Press Inc, Stamford, 1992 Search PubMed.
- A. Ono and H. Togashi, Angew. Chem., Int. Ed., 2004, 43(33), 4300–4302 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46(40), 7587–7590 CrossRef CAS PubMed.
- C. K. Chiang, C. C. Huang, C. W. Liu and H. T. Chang, Anal. Chem., 2008, 80(10), 3716–3721 CrossRef CAS PubMed.
- J. Lee, M. S. Han and C. A. Mirkin, Angew. Chem., Int. Ed., 2007, 46(22), 4093–4096 CrossRef CAS PubMed.
- D. Li, A. Wieckowska and I. Willner, Angew. Chem., Int. Ed., 2008, 47(21), 3927–3931 CrossRef CAS PubMed.
- S. J. Liu, H. G. Nie, J. H. Jiang, G. L. Shen and R. Q. Yu, Anal. Chem., 2009, 81(14), 5724–5730 CrossRef CAS PubMed.
- G. Mor-Piperberg, R. Tel-Vered, J. Elbaz and I. Willner, J. Am. Chem. Soc., 2010, 132(20), 6878–6879 CrossRef CAS PubMed.
- G. Wang, X. He, B. Wang, X. Zhang and L. Wang, Analyst, 2012, 137(9), 2036–2038 RSC.
- M. Kumar and P. Zhang, Biosens. Bioelectron., 2010, 25(11), 2431–2435 CrossRef CAS PubMed.
- B. Zhang and L. H. Guo, Biosens. Bioelectron., 2012, 37(1), 112–115 CrossRef CAS PubMed.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21(10), 1192–1199 CrossRef CAS PubMed.
- Y. Zhang, G. M. Zeng, L. Tang, D. L. Huang, X. Y. Jiang and Y. N. Chen, Biosens. Bioelectron., 2007, 22(9–10), 2121–2126 CrossRef CAS PubMed.
- L. Tang, G. M. Zeng, G. L. Shen, Y. P. Li, Y. Zhang and D. L. Huang, Environ. Sci. Technol., 2008, 42(4), 1207–1212 CrossRef CAS.
- C. I. L. Justino, T. A. P. Rocha-Santos, S. Cardoso and A. C. Duarte, TrAC, Trends Anal. Chem., 2013, 47(7), 27–36 CrossRef CAS PubMed.
- H. W. Ra, J. T. Kim, R. Khan, D. Sharma, Y. G. Yook, Y. B. Hahn, J. H. Park, D. G. Kim and Y. H. Im, Nano Lett., 2012, 12(4), 1891–1897 CrossRef CAS PubMed.
- Y. Li, H. J. Schluesener and S. Xu, Gold Bull., 2010, 43(1), 29–41 CrossRef CAS.
- Y. Zhang, G. M. Zeng, L. Tang, Y. P. Li, L. J. Chen, Y. Pang, Z. Li, C. L. Feng and G. H. Huang, Analyst, 2011, 126(20), 4204–4210 RSC.
- W. Zhao, J. J. Xu, C. G. Shi and H. Y. Chen, Electrochem. Commun., 2006, 8(5), 773–778 CrossRef CAS PubMed.
- L. Chen, G. Zeng, Y. Zhang, L. Tang, D. Huang, C. Liu, Y. Pang and J. Luo, Anal. Biochem., 2010, 407(2), 172–179 CrossRef CAS PubMed.
- E. L. S. Wong, P. Erohkin and J. J. Gooding, Electroanal. Chem., 2004, 6(7), 648–654 CAS.
- S. Manghani and J. J. Ramsden, J. Biol. Phys. Chem., 2003, 3(1), 11–17 CrossRef CAS.
- B. A. D. Neto and A. A. M. Lapis, Molecules, 2009, 14(5), 1725–1746 CrossRef CAS PubMed.
- D. Wu, Q. Zhang, X. Chu, H. Wang, G. Shen and R. Yu, Biosens. Bioelectron., 2010, 25(5), 1025–1031 CrossRef CAS PubMed.
- R. M. Kong, X. B. Zhang, L. L. Zhang, X. Y. Jin, S. Y. Huan, G. L. Shen and R. Q. Yu, Chem. Commun., 2009, 5633–5635 RSC.
- Z. Zhu, Y. Su, J. Li, D. Li, J. Zhang, S. Song, Y. Zhao, G. Li and C. Fan, Anal. Chem., 2009, 81(18), 7660–7666 CrossRef CAS PubMed.
- X. Zhang, Y. Li, H. Su and S. Zhang, Biosens. Bioelectron., 2010, 25(6), 1338–1343 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47871h |
| This journal is © The Royal Society of Chemistry 2014 |