An efficient facile and one-pot synthesis of benzodiazepines and chemoselective 1,2-disubstituted benzimidazoles using a magnetically retrievable Fe3O4 nanocatalyst under solvent free conditions†
Ramen Jamatia,
Mithu Saha and
Amarta Kumar Pal*
Department of Chemistry, North Eastern Hill University, Mawlai Campus, Shillong 793022, India. E-mail: amartya_pal22@yahoo.com; Fax: +91 364 2550076; Tel: +91 364 2307930 ext. 2636
First published on 21st February 2014
Abstract
Benzodiazepine and chemoselective 1,2-disubstituted benzimidazole derivatives were synthesized by the condensation reaction of o-phenylenediamine with ketones and aryl aldehydes using Fe3O4 nanoparticles as a recyclable catalyst under solvent free conditions. This synthetic approach eliminates the use of toxic organic solvents with the added benefit of easy separation and reusability of the catalyst without compromising the yield or purity which makes the procedure green.
Green chemistry is the development of reaction processes that reduce or eliminate the use or generation of hazardous substances.1 In the last decade emphasis has been given to developing more environmentally benign synthetic organic processes. Organic solvents pose the biggest challenge in this regard due to their hazardous impact on the environment and the human health. The design of a reaction process without the use of hazardous organic solvent would be an important step towards green chemistry. The present approach offers the advantage of eliminating the use of hazardous organic solvents.
Recently, metal nanoparticles have received much attention in the field of organic synthesis. Metal nanoparticles are much more reactive than the bulk because of their higher surface to volume ratio.2 Further, the reusability of the catalyst has become an important trend in chemistry due to growing environmental and economic concern. Of the nanoparticles, Fe3O4 NPs are important because of their potential uses such as in magnetic drug targeting, clinical diagnosis and as catalyst.3 Fe3O4 NPs, are also of interest due to its easy synthesis and magnetic property which makes it easily separable by an external magnetic field and being comparatively cheap.
Nitrogen containing [6, 7] and [6, 5] fused heterocycles like benzodiazepine and benzimidazole derivatives are important class of heterocyclic compounds having interesting pharmacological and biological properties (Fig. 1).4,5,30–32 These compounds have also been known for their analgesic, antianxiety, hypnotic, anti-inflammatory, anticonvulsant, muscle-relaxant, antitumor, antiulcer, antimicrobial, antiviral, anti-HIV and amnesic properties.6–9 Benzimidazole has also been found to be effective against human cytomegalovirus (HCMV) and as efficient selective neuropeptide YY1 receptor antagonists.10,11 Because of these medicinal and industrial applications, these compounds have been of wide interest. These compounds can also be used for the preparation of other important heterocyclic compounds and are important intermediates in many organic reactions.12–15
Benzodiazepines and benzimidazole derivatives have been prepared by the condensation or cyclization of o-phenylenediamine with a variety of carbonyl compounds. Various catalyst such as BF3·Et2O,16 NBS,17 NaBH4,18 LaCl3·7H2O,19 polyphosphoric acid,20 AgNO3,21 Yb(OTf)3,22 Sc(OTf)3,23 Ga(OTf)3,24 ZnCl2,25 and ionic liquids,26 have been employed for the synthesis of benzodiazepine. Benzimidazole have also been reported with amberlite,27 Fe(ClO4)3,28 In2O3.29 However these methods suffer from drawbacks such as the use of hazardous organic solvents, longer reaction time, low yield, harsher reaction condition and high cost. In our present approach, we have synthesized benzodiazepine and benzimidazole derivatives under solvent free condition using Fe3O4 NPs which eliminates the use of toxic solvents with the added advantage of reusability of the catalyst.
 | ||
| Fig. 1 Some biologically important compounds. | ||
Results and discussion
Fe3O4 NPs was prepared by the addition of a base to an aqueous solution containing Fe3+ and Fe2+ in a molar ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, according to the procedure given by Karami et al.33 The resulting solution was heated at 50–60 °C with stirring. A dark black precipitate was formed. Then the precipitate was filtered, dried and characterized by TEM, SEM, XRD, EDAX, UV and IR. The equation can be expressed as
:1, according to the procedure given by Karami et al.33 The resulting solution was heated at 50–60 °C with stirring. A dark black precipitate was formed. Then the precipitate was filtered, dried and characterized by TEM, SEM, XRD, EDAX, UV and IR. The equation can be expressed as| Fe2+ + 2Fe3+ + 8HO− → Fe3O4 + 4H2O |
XRD patterns of Fe3O4 NPs are shown in Fig. 2. The XRD patterns shows a number of prominent Bragg reflections by their indices (220), (311), (400), (422), (511) and (440) which indicates that the resultant nanoparticles were Fe3O4 with a spinel structure. The broad peak is an indication that the particles were of nanoscale size. The size of the particles was examined by transmission electron microscope (TEM) and the TEM image (Fig. 3a) clearly shows a monodispersed spherical shaped Fe3O4 NPs. The morphology and particle size of the Fe3O4 NPs was studied using SEM. The SEM image (Fig. 4a) indicates that the Fe3O4 NPs are spherical in shape and in the nanometer range. Characterization of Fe3O4 NPs was also done using EDAX. The EDAX spectra show a strong peak of Fe (Fig. 5). The characterization of the Fe3O4 NPs was also studied using UV and IR. The UV spectra show a characteristic absorption bands at 370 nm which corresponds to the Fe3O4 NPs (Fig. 6), which originate primarily from the absorption and scattering of UV radiation by magnetic nanoparticles. The results was found to be in good agreement with those reported in the literature.34 The particles size were found to be 10–20 nm before used. The distributions of Fe3O4 NPs are shown in Fig. 7. The IR spectrum shows bands at 599 cm−1 and 3430 cm−1 which indicates Fe–O structure and OH group for spinel Fe3O4 (Fig. S.I-1†).35
 | ||
| Fig. 2 Powder XRD of Fe3O4 NPs. | ||
 | ||
| Fig. 3 TEM image of Fe3O4 NPs before and after use. | ||
 | ||
| Fig. 4 SEM image of Fe3O4 NPs before and after use. | ||
 | ||
| Fig. 5 EDAX spectra of Fe3O4 NPs. | ||
 | ||
| Fig. 6 UV visible spectra of Fe3O4 NPs. | ||
 | ||
| Fig. 7 Histogram diagram of Fe3O4 NPs. | ||
The synthesized Fe3O4 NPs was then applied as a reusable catalyst in a condensation reaction which intern will produce biologically important heterocyclic compounds such as benzodiazepine and their derivatives. The model condensation reaction between o-phenylenediamine (1, 1 mmol) and acetophenone (2, 2.2 mmol) in presence of Fe3O4 NPs was carried out under solvent free condition (Scheme 1). The reaction was carried out in various solvents, like water, THF, acetonitrile, ethanol and toluene. Significant improvement was achieved in ethanol and acetonitrile, but the best result was observed under solvent free condition (Fig. 8). However, the reaction did not proceed in water medium which might be due to poor solubility of the starting materials.
 | ||
| Scheme 1 | ||
 | ||
| Fig. 8 Chart for the optimization of solvent effect. | ||
The amount of catalyst concentration for the model reaction was scanned. Firstly, the condensation reaction of o-phenylenediamine (1, 1 mmol) and acetophenone (2, 2.2 mmol) was carried out in absence of catalyst, very less conversion was observed (22%). The reaction was then studied with various mol% of the catalyst (2–10 mol%). It was found that the product yield proportionally increased with catalyst concentration. Maximum yield was obtained by using 6 mol% of the catalyst. Further increase in the catalyst concentration (8 mol% and 10 mol%), the yield of the product did not improve. So 6 mol% is the optimum catalyst concentration is this reaction (Fig. 9).
 | ||
| Fig. 9 Effect of catalyst loading on the reaction yield. | ||
Rationalising the above results, we carried out the said condensation reaction of o-phenylenediamine (1, 1 mmol) and acetophenone (2, 2.2 mmol) in presence of Fe3O4 NPs (6 mol%) under solvent free condition at 80 °C (Scheme 1). The reaction went to completion within 15 min yielding a solid pale white product in high yield (89%). The structure of the compound was established by analytical and spectroscopic methods. The presence of peaks in 1H NMR at 3.4 (brs, 1H), 3.08 (d, J = 13.2 Hz, 1H) and 2.92 (d, J = 13.2 Hz, 1H) due to NH and methylene protons and peak at 3281 cm−1 due to NH stretching in IR spectra clearly indicates the formation of 3a. The effect of temperature on the product yield was also studied. The reaction was carried out at room temperature but no desired product was obtained only starting materials were recovered after 1 h. With increase in the temperature, yield of the desired product increases and maximum yield was achieved at 80 °C (Table 1).
| Carbonyl compound | Product | Time (min) | Yielda (%) | Mp (°C) [found] | Mp (°C) [lit.] | |
|---|---|---|---|---|---|---|
| a Isolated yields.b NF = not found. | ||||||
| 1 |  |
 |
15 | 89 | 150–152 | [151–152]22 |
| 2 |  |
 |
12 | 93 | 137–139 | [147–149]24 |
| 3 | 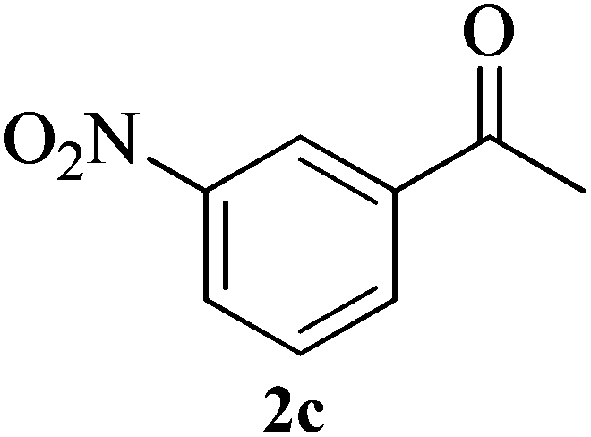 |
 |
10 | 90 | 154–156 | [164–166]24 |
| 4 |  |
 |
14 | 91 | 135–137 | [145–146]19 |
| 5 |  |
 |
10 | 95 | 152–154 | [154–155]24 |
| 6 |  |
 |
15 | 88 | 97–100 | [99–101]24 |
| 7 |  |
 |
15 | 89 | 113–116 | [115–116]24 |
| 8 | 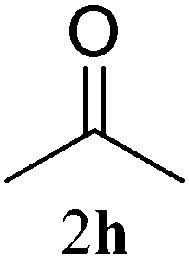 |
 |
13 | 89 | 133–135 | [137–139]22 |
| 9 |  |
 |
15 | 87 | 129–130 | [134]28 |
| 10 |  |
 |
10 | 91 | 131–135 | [137]28 |
| 11 |  |
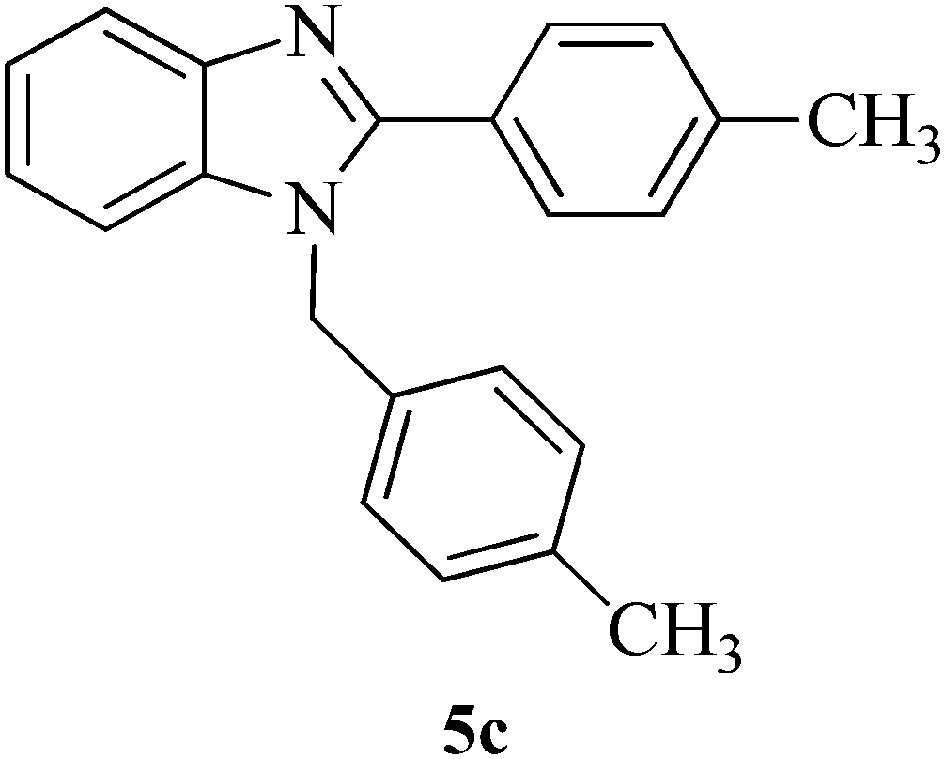 |
10 | 89 | 125–127 | [125]28 |
| 12 | 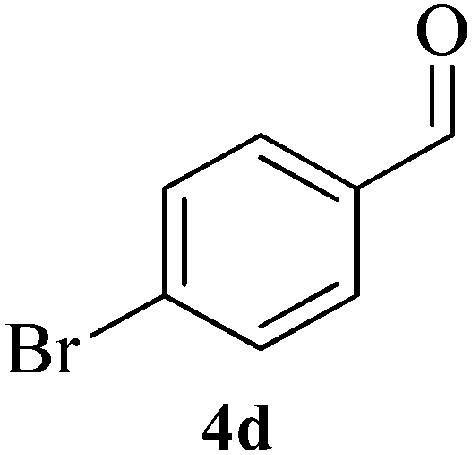 |
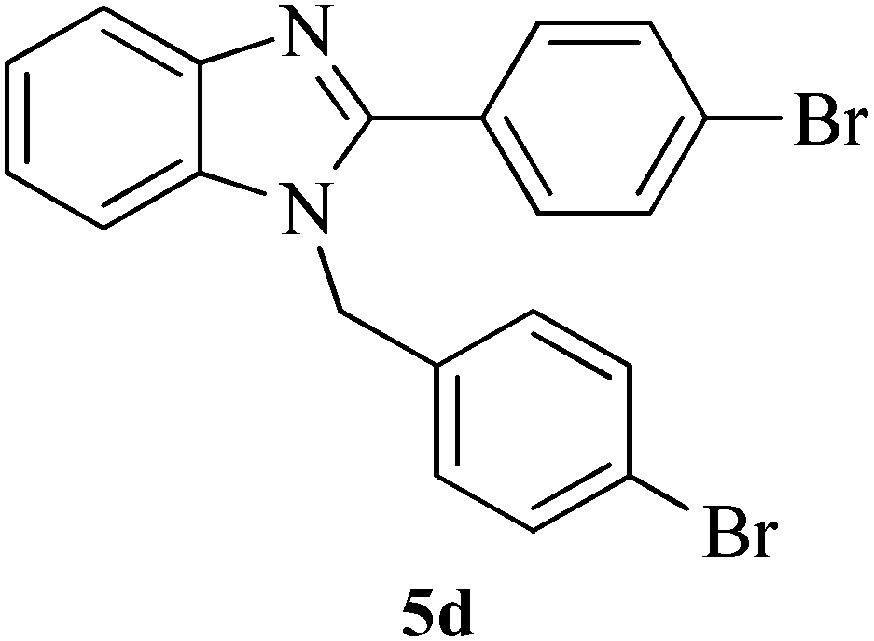 |
15 | 92 | 157–159 | [157–158]27 |
| 13 |  |
 |
15 | 86 | 116–119 | NFb |
| 14 |  |
 |
10 | 90 | 149–150 | [150]28 |
To make the process more general, the model reaction was carried out with different substituted ketones. In all the cases, the reactions were completed within very short period and furnished the corresponding products (3a–h) in higher yield (Table 1).
Encouraged by the initial success, next we decided to explore the synthesis of biologically important benzimidazole and their derivatives using the present protocol. To our delight, it was observed that the reaction worked well and furnished chemoselectively the 1,2-disubstituted benzimidazoles (5a–f) instead of a mixture of monosubstituted and disubstituted product irrespective of the molar ratio used (Scheme 1).36 We also carried out the reaction using various aryl aldehydes having electron donating or electron withdrawing groups. In all the cases, desired product was achieved in good yields (Table 1).
The plausible mechanism for the formation of compounds 3a–h and 5a–f is given below (Scheme 2). Initially the Fe3O4 NPs facilitated the reaction between diamine 1 and ketones 2 or aldehyde 4, which generates the common intermediate 9. The intermediate 9 undergoes tautomerism to form intermediate 10. The intermediate 10 then undergoes intramolecular cyclization followed by hydride shift to furnish the final benzodiazepine products 3a–h.37 In case of aryl aldehydes, intermediate 9 undergoes cyclization (12) followed by 1,3-hydride shift to produce the disubstituted benzimidazole products 5a–f.38
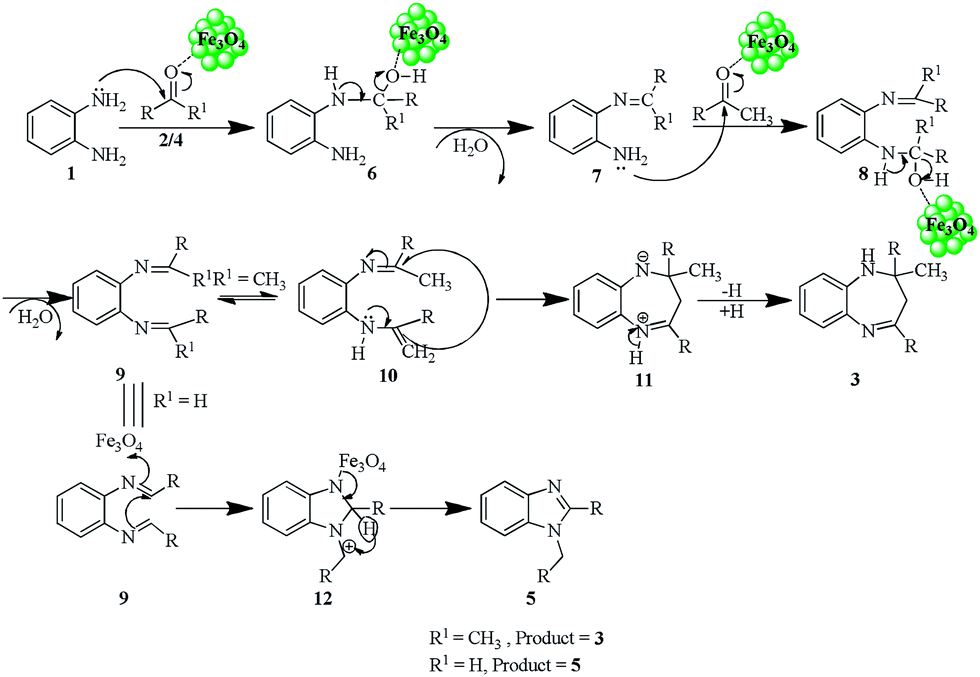 | ||
| Scheme 2 | ||
Reusability is one of the most important properties of a good catalyst. So, we checked the reusability of the catalyst in our present protocol. After the completion of the reaction, the reaction mixture was dissolved in 10 mL of ethyl acetate and the catalyst was separated using external magnetic field. The separated catalyst was then dried and reused for another set of reaction. To our excitement, it was found that the Fe3O4 NPs can be reused for four consecutive runs without any appreciable decrease in its catalytic activity (Fig. 10).
 | ||
| Fig. 10 Reusability chart. | ||
Structure of the compound 3b was further confirmed by X-ray crystallography. The compound 3b was carefully recrystallized from ethanol. Fig. 11 shows the ORTEP diagram of compound 3b.†

Conclusions
In conclusion, we have developed a new efficient and simple method for the synthesis of benzodiazepine and chemoselective 1,2-disubstituted benzimidazole derivatives. This method will be very useful in industrial point of view because it is high yielding, solvent free and catalyst can be separated by external magnetic field and reused for several runs.Experimental
Melting points were determined in open capillaries and are uncorrected. IR spectra were recorded on Spectrum BX FT-IR, Perkin Elmer (υmax in cm−1) on KBr disks.1H NMR and 13C NMR (400/300 MHz and 100/75 MHz respectively) spectra were recorded on Bruker Avance II-400 and 300 spectrometer in CDCl3 (chemical shifts in δ with TMS as internal standard). Mass spectra were recorded on Waters ZQ-4000. Transmission Electron Microscope (TEM) was recorded on JEOL JSM 100CX. Scanning electron microscope (SEM) was recorded on JSM-6360 (JEOL). XRD was recorded on Bruker D8 XRD instrument SWAX.CHN were recorded on CHN-OS analyzer (Perkin Elmer 2400, Series II). Silica gel G (E-mark, India) was used for TLC. Hexane refers to the fraction boiling between 60 °C and 80 °C. Absorption spectra were recorded in Lambda25 (Perkin Elmer Inc.) spectrometers.X-ray crystallography
The X-ray diffraction data were collected at 293 K with Mo Kα radiation (λ = 0.71073 Å) using Agilent Xcalibur (Eos, Gemini) diffractometer equipped with a graphite monochromator. The software used for data collection CrysAlis PRO (Agilent, 2011), data reduction CrysAlis PRO and cell refinement CrysAlis PRO. The structure were solved by direct methods and refined by full-matrix least-squares calculation using SHELXS-9739 and SHELXL-97.40†General procedure for the synthesis of 3a–h
Fe3O4 (6 mol%) was added to a mixture of o-phenylenediamine 1 (1 mmol) and acetophenone 2a–h (2.2 mmol) and heated at 80 °C under solvent free condition. After completion of the reaction, monitored by TLC, the reaction mixture was cooled to room temperature and dissolved in 10 mL ethyl acetate. The Fe3O4 NPs was then separated by external magnetic field. The separated Fe3O4 NPs was washed with ethyl acetate and dried. Then it was used for another set of reaction under similar condition. The reaction mixture was then washed with water (3 × 5 mL), brine (1 × 5 mL) and dried over anhydrous Na2SO4. The reaction mass was concentrated under vacuum and the pure product was obtained by purification through column chromatography using ethyl acetate–hexane as eluent.General procedure for the synthesis of 5a–f
In a round bottom flask, o-phenylenediamine 1 (1 mmol) and aldehyde 4a–f (2.2 mmol), was taken and Fe3O4 NPs (6 mol%) was added. The reaction was carried out at 80 °C under solvent free condition. After completion (TLC), the reaction mixture was cooled to room temperature and dissolved in 10 mL ethyl acetate. The Fe3O4 NPs was then separated by external magnetic field and washed with ethyl acetate and dried. The separated Fe3O4 NPs was then used for another set of reaction under similar condition. The reaction mixture was then washed with water (3 × 5 mL), brine (1 × 5 mL) and dried over anhydrous Na2SO4. The reaction mass was concentrated under vacuum. The crude mixture was purified by column chromatography using ethyl acetate–hexane as eluent.Spectral data for selected compounds
Compound 3b: (entry 2): yellow solid. IR (KBr): 3496, 3025, 1686, 1493, 751 cm−1. 1H NMR (CDCl3, 300 MHz): δ = 7.51–7.45 (m, 4H), 7.30–7.25 (m, 1H), 7.20–7.17 (m, 4H), 7.07–7.01 (m, 2H), 6.82–6.76 (m, 1H), 3.41 (s, 1H), 3.07 (d, J = 13.3 Hz, 1H), 2.88 (d, J = 13.5, 1H), 1.71 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ = 166.1, 145.7, 139.8, 137.6, 136.1, 133.0, 128.5, 128.38, 128.31, 127.0, 126.6, 122.0, 121.5, 73.5, 42.9, 29.7. ESI-MS: m/z 381, 383 [M + H]+. Anal. calcd for C22H18Cl2N2: C, 69.30; H, 4.76; N, 7.35. Found: C, 69.53; H, 4.64; N, 7.44%.Compound 3c: (entry 3): orange solid. IR (KBr): 3278, 3088, 1604, 1473, 1349 cm−1. 1H NMR (CDCl3, 300 MHz): δ = 8.47 (s, 1H), 8.16 (s, 1H), 8.12 (d, J = 8.1 Hz, 1H), 7.99–7.96 (m, 3H), 7.42–7.32 (m, 3H), 7.18–7.08 (m, 2H), 6.93–6.91 (dd, J = 7.5 Hz, 1.2 Hz, 1H), 3.55 (s, 1H), 3.28 (d, J = 13.5 Hz, 1H), 3.02 (d, J = 13.5, 1H), 1.87 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ = 164.1, 149.0, 148.1, 148.0, 140.4, 139.2, 137.1, 132.5, 131.9, 129.5, 129.2, 128.9, 127.4, 124.4, 122.4, 122.2, 121.6, 120.8, 74.1, 42.8, 29.9. ESI-MS: m/z 403 [M + H]+. Anal. calcd for C22H18N4O4: C, 65.66; H, 4.51; N, 13.92. Found: C, 65.61; H, 4.62; N, 13.80%.
Compound 3d: (entry 4): pale brown solid. IR (KBr): 3370, 2979 cm−1. 1H NMR (CDCl3, 300 MHz): δ = 7.46–7.27 (m, 9H), 7.11–7.01 (m, 2H), 6.83–6.80 (m, 1H), 3.42 (s, 1H), 3.07 (d, J = 13.2 Hz, 1H), 2.89 (d, J = 13.2 Hz, 1H), 1.72 (s, 3H). 13C NMR (CDCl3, 100 MHz): δ = 166.2, 146.5, 140.0, 138.3, 137.7, 131.5, 131.4, 128.8, 128.7, 127.6, 126.8, 124.7, 122.2, 121.7, 121.4, 73.7, 43.0, 29.9. ESI-MS: m/z 469, 471 [M + H]+. Anal. calcd for C22H18Br2N2: C, 56.20; H, 3.86; N, 5.96. Found: C, 56.12; H, 3.88; N, 5.77%.
Compound 5b: (entry 10): pale white solid. IR (KBr): 3075, 2928, 2852, 1611, 1250, 744 cm−1. 1H NMR (CDCl3, 400 MHz): δ = 7.81 (d, J = 7.6 Hz, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 7.29–7.19 (m, 4H), 7.14 (d, J = 8 Hz, 1H), 6.96 (d, J = 8 Hz, 2H), 5.33 (s, 2H). 13C NMR (CDCl3, 100 MHz): δ = 152.8, 142.9, 136.3, 135.8, 134.6, 133.8, 130.4, 129.3, 129.1, 128.2, 127.2, 123.5, 123.1, 120.1, 110.3, 47.8. ESI-MS: m/z 353, 355 [M + H]+. Anal. calcd for C20H14 Cl2N2: C, 68.00; H, 3.99; N, 7.93. Found: C, 68.21; H, 4.03; N, 7.82%.
Acknowledgements
We thank the Department of Chemistry, Sophisticated Analytical and Instrumentation Facility (SAIF) of North-Eastern Hill University, SAIF-CDRI Lucknow, UGC for supporting this work under Special Assistance Programme (SAP) and DST-Purse programme of NEHU Shillong. We are also thankful to DST for financial support (sanctioned no: SERC/F/0293/2012-13), Dr S. Khatua and Dr S. L. Nongbri.Notes and references
- P. T. Anastas, L. G. Heine and T. C. Williamson, Green Chemical Syntheses and Processes: Introduction, 2000, vol. 1, p. 1 Search PubMed.
- C. Burda, X. Chen, R. Narayana and M. A. E. Sayed, Chem. Rev., 2005, 105, 1025 CrossRef CAS PubMed.
- (a) S. Lian, E. Wang, Z. Kang, Y. Bai, L. Gao, M. Jiang, C. Hu and L. Xu, Solid State Commun., 2004, 129, 485 CrossRef CAS PubMed; (b) V. S. Zaitsev, D. S. Filimonov, I. A. Presnyakov, R. J. Gambino and B. Chu, J. Colloid Interface Sci., 1999, 212, 49 CrossRef CAS PubMed.
- H. Schutz, Benzodiazepines, Springer, Heidelberg, 1982, vol. 2, p. 240 Search PubMed; J. K. Landquist, in Comprehensive Heterocyclic Chemistry, ed. A. R. Katritzky and C. W. Rees, Pergamon Press, Oxford, U.K., 1984, vol. 1, p. 166 Search PubMed.
- (a) H. Nakano, T. Inoue, N. Kawasaki, H. Miyataka, H. Matsumoto, T. Taguchi, N. Inagaki, H. Nagai and T. Satoh, Bioorg. Med. Chem., 2000, 8, 373 CrossRef CAS; (b) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893 CrossRef CAS PubMed.
- L. O. Randall and B. Kappel, Benzodiazepines, ed. S. Garattini, E. Mussini and L.O. Randall, Raven Press, New York, 1973, p. 27 Search PubMed.
- J. R. De Baun, F. M. Pallos and D. R. Baker, US Pat. 3, 1976, vol. 978, p. 227, Chem. Abstr., 1977, 86, 5498.
- W. A. Denny, G. W. Rewcastle and B. C. Bagley, J. Med. Chem., 1990, 33, 814 CrossRef CAS.
- T. Fonseca, B. Gigante and T. L. Gilchrist, Tetrahedron, 2001, 57, 1793 CrossRef CAS.
- Z. Zhu, B. Lippa, J. C. Drach and L. B. Townsend, J. Med. Chem., 2000, 43, 2430 CrossRef CAS PubMed.
- H. Zarrinmayeh, D. M. Zimmerman, B. E. Cantrell, D. A. Schober and R. F. Bruns, Bioorg. Med. Chem. Lett., 1999, 9, 647 CrossRef CAS.
- M. Essaber, A. Baouid, A. Hasnaoui, A. Benharref and J. P. Lavergne, Synth. Commun., 1998, 28, 4097 CrossRef CAS.
- (a) J. X. Xu, H. T. Wu and S. Jin, Chin. J. Chem., 1999, 17, 84 CrossRef CAS; (b) X. Y. Zhang, J. X. Xu and S. Jin, Chin. J. Chem., 1999, 17, 404 CrossRef CAS.
- K. V. V. Reddy, P. S. Rao and D. Ashok, Synth. Commun., 2000, 30, 1825 CrossRef CAS.
- Y. Bai, J. Lu, Z. Shi and B. Yang, Synlett, 2001, 4, 544 Search PubMed.
- J. A. L. Herbert and H. Suschitzky, J. Chem. Soc., Perkin Trans. 1, 1974, 1, 2657 RSC.
- C.-K. Kuo, S. V. More and C.-F. Yao, Tetrahedron Lett., 2006, 47, 8523 CrossRef CAS PubMed.
- H. R. Morales, A. Bulbarela and R. Contreras, Heterocycles, 1986, 24, 135 CrossRef CAS.
- S. S. Pandit, B. D. Vikhe and G. D. Shelke, J. Chem. Sci., 2007, 119, 295 CrossRef CAS.
- D. I. Jung, T. W. Choi, Y. Y. Kim, I. S. Kim, Y. M. Park, Y. G. Lee and D. H. Jung, Synth. Commun., 1999, 29, 1941 CrossRef CAS.
- R. Kumar, P. Chaudhary, S. Nimesh, A. K. Verma and R. Chandra, Green Chem., 2006, 8, 519 RSC.
- M. Curini, F. Epifano, M. C. Marcotullio and O. Rosati, Tetrahedron Lett., 2001, 42, 3193 CrossRef CAS.
- S. K. De and R. A. Gibbs, Tetrahedron Lett., 2005, 46, 1811 CrossRef CAS PubMed.
- X.-Q. Pan, J.-P. Zou, Z.-H. Huang and W. Zhang, Tetrahedron Lett., 2008, 49, 5302 CrossRef CAS PubMed.
- M. A. Pasha and V. P. Jayashankara, Heterocycles, 2006, 68, 1017 CrossRef CAS.
- D. V. Jarikote, S. A. Siddiqui, R. Rajagopal, D. Thomas, R. J. Lahoti and K. V. Srinivasan, Tetrahedron Lett., 2003, 44, 1835 CrossRef CAS.
- S. D. Sharma and D. Konwar, Synth. Commun., 2009, 39, 980 CrossRef.
- H. A. Oskooie, M. M. Heravi, A. Sadnia, F. K. Behbahani and F. Jannati, Chin. Chem. Lett., 2007, 18, 1357 CrossRef CAS PubMed.
- S. Santra, A. Majee and A. Hajra, Tetrahedron Lett., 2012, 53, 1974 CrossRef CAS PubMed.
- M. E. Tranquillini, P. G. Cassara, M. Corsi, G. Curotto, D. Donati, G. Finizia, G. Pentassuglia, S. Polinelli, G. Tarzia, A. Ursini and F. T. M. V. Amsterdam, Arch. Pharm., 1997, 330, 353 CrossRef CAS.
- D. A. Claremon, N. Liverton, H. G. Selnick and G. R. Smith, PCT Int. Appl. WO 9640653, 2003.
- M. D. Rosen, Z. M. Simon, K. T. Tarantino, X. L. Zhao and M. H. Rabinowitz, Tetrahedron Lett., 2009, 50, 1219 CrossRef CAS PubMed.
- B. Karami, S. J. Hoseini, K. Eskandari, A. Ghasemib and H. Nasrabadia, Catal. Sci. Technol., 2012, 2, 331 CAS.
- O. U. Rahman, S. C. Mohapatra and S. Ahmad, Mater. Chem. Phys., 2012, 32, 196 CrossRef PubMed.
- Y.-P. Chang, C.-L. Ren, J.-L. Qu and X.-G. Chen, Appl. Surf. Sci., 2012, 61, 504 CrossRef PubMed.
- V. Kumar, D. G. Khandare, A. Chatterjee and M. Banerjee, Tetrahedron Lett., 2013, 54, 5505 CrossRef CAS PubMed.
- (a) S. K. De and R. A. Gibbs, Tetrahedron Lett., 2005, 46, 1811 CrossRef CAS PubMed; (b) M. Saha, A. K. Pal and S. Nandi, RSC Adv., 2012, 2, 6397 RSC.
- J.-P. Wan, S.-F. Gan, J.-M. Wu and Y. Pan, Green Chem., 2009, 11, 1633 RSC.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 96, 467 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 645, 112 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 933681. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47860b |
| This journal is © The Royal Society of Chemistry 2014 |