 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanism of ionic Diels–Alder reactions. A DFT study of the oxa-Povarov reaction†
Luis R. Domingo*a,
Maria J. Aurella and
Patricia Pérez*b
aUniversidad de Valencia, Departamento de Química Orgánica, Dr Moliner 50, E-46100 Burjassot, Valencia, Spain. E-mail: domingo@utopia.uv.es; Web: www.luisrdomingo.com
bUniversidad Andrés Bello, Facultad de Ciencias Exactas, Departamento de Ciencias Químicas, Laboratorio de Química Teórica, Av. República 275, 8370146 Santiago, Chile
First published on 21st March 2014
Abstract
The mechanism of the oxa-Povarov reaction of a cationic aryl oxonium with cyclopentene and styrene has been studied using DFT methods at the B3LYP/6-31G* level as a reaction model of ionic Diels–Alder (I-DA) reactions. Oxa-Povarov reactions are initialized by an I-DA reaction between the cationic aryl oxonium and alkenes to yield the corresponding formal [4+ + 2] cycloadducts. The last step is a rapid loss of a proton giving chromans. While the I-DA reaction with cyclopentene takes place through a two-stage one-step mechanism, the presence of a phenyl substituent on styrene makes the mechanism of the I-DA reaction stepwise. An electron localization function (ELF) bonding analysis of selected points along the IRCs of these I-DA reactions allows the establishment of a great similarity in bond formation under both one-step and two-step mechanisms. The formation of the first C–C single bond begins in the short range of 1.95–1.90 Å, by the coupling of two pseudoradical centers generated at the most electrophilic center of the cationic aryl oxonium and the most nucleophilic centers of cyclopentene and styrene, resulting in the global charge transfer that takes place during I-DA reactions. The use of the recently proposed radical Pok Parr functions allows the characterization of the most electrophilic centers in cationic species and the most nucleophilic centers in anionic species.
Introduction
The Diels–Alder (DA) reaction is arguably one of the most powerful reactions in the great arsenal of synthetic organic chemistry.1 By varying the nature of the diene and dienophile, many different types of carbocyclic structures can be built with high stereoselectivity and total regioselectivity. An exhaustive study of DA reactions allowed us to find good correlations between the experimental reaction rates and the global charge transfer (GCT) computed at the transition state structures (TS) of the reactions.2 This finding allowed us, in 2009, to propose the polar Diels–Alder (P-DA) reaction mechanism (see Scheme 1). This mechanism is characterized by favorable nucleophilic/electrophilic interactions at the TSs, instead of molecular orbital interactions as proposed by the FMO theory.3 Only few DA reactions do not follow the polar mechanism. However, these non-polar Diels–Alder (N-DA) reactions, which are characterized by a very low GCT at the TSs, are of low synthetic interest due to the drastic reaction conditions needed for the reaction to take place. | ||
| Scheme 1 | ||
In general, in a DA reaction, a neutral conjugated diene reacts with a neutral ethylene derivative to yield a six-membered carbocyclic system. However, in some occasions, positively or negatively charged ionic species can participate in DA reactions. In such cases, the ionic nature of the reaction is preserved along the reaction until formation of the ionic cycloadduct (CA) (see Scheme 1).2 In these cases, although a large GCT also takes place along the reaction, they must to be classified differently from P-DA reactions, since the zwitterionic character of the latter is evidenced only along the reaction, as reagents and CAs are neutral.
This behavior prompted us to establish the ionic Diels–Alder (I-DA) reaction type, in which reagents, TSs, feasible intermediates, and CAs remain charged during the cycloaddition reaction.2 I-DA reactions can be classified as anionic and cationic DA reactions. However, while cationic DA reactions take place quickly at very low temperatures,4 usually at −78 °C, due to the high electrophilic character of cationic species, there are few anionic DA reactions because, in spite of the high nucleophilic character of anionic species, these reactions do not take place easily in absence of strong electrophiles.5
Several theoretical studies have been devoted to I-DA reactions.6–9 Thus, TS associated with the I-DA reaction of N,N-dimethyliminium cation 1 with Cp 2 (ref. 9) was found to be energetically below the reagents as a consequence of the strong electrophilic character of 1, ω = 8.42 eV (see Scheme 2).6 However, when the formation of a molecular complex MC1 was considered, the activation energy of this I-DA reaction became positive. This I-DA reaction, which takes place via highly asynchronous TS, was associated with the nucleophilic attack of Cp 2 on the carbon atom of 1, followed by a concomitant ring closure, yielding [4 + 2+] CA 3.6
 | ||
| Scheme 2 | ||
On the other hand, the I-DA reaction of diprotonated iminium cation 4, ω = 22.79 eV, with Cp 2 (ref. 10) presented a two-step mechanism with formation of cationic intermediate IN (see Scheme 3).7 Similar to the I-DA reactions of 1 with Cp 2, TS1 was found to be −17 kcal mol−1 below the reagents. Only when the formation of MC2 was considered, the activation energy became slightly positive.
 | ||
| Scheme 3 | ||
A comparative analysis of the geometric and electronic parameters of TS and TS1 shows that both structures present a large similarity in the C–C single bond formation. In spite of this, while IRC calculations from TS1 finish at the cationic intermediate IN, IRC calculations from TS end at the formal [4 + 2+] CA 3. An exhaustive analysis of the corresponding IRC shows that the formation of the two single bonds in this one-step process takes place through a two-stage mechanism;11 i.e. while the C–C bond formation takes place at the first stage of the reaction along the nucleophilic attack of Cp 2 on iminium cation 1, the second C–N single bond is formed along a ring closure process at the second stage of the reaction, yielding [4 + 2+] CA 3.
Very recently, Batey et al. have reported the synthesis of chromans such as 9 and 14, via the oxa-Povarov reaction of the oxonium ion 6 with several alkenes, including cyclopentene 7 and styrene 12 (see Scheme 4).12 These oxa-Povarov reactions begin by one I-DA reaction of oxonium ion 6 with alkenes 7 and 12 yielding the formal [4+ + 2] CAs 8 and 13, which by one rapid loss of a proton afford chromans 9 and 14. These reactions presented endo selectivity, while in the cases of the asymmetric styrene 12, the reaction was completely regioselective.
 | ||
| Scheme 4 | ||
For the I-DA reactions, the authors proposed two competitive mechanisms:12 (i) a direct asynchronous [4+ + 2] cycloaddition pathway, and (ii) a stepwise process initialized by the Prins addition of alkenes to the aryl 2-oxadiene oxonium 6, followed by an intramolecular aromatic electrophilic substitution reaction. The authors carried out a series of experiments in order to establish the mechanism of these I-DA reactions. Thus, the reaction of cis-β-methylstyrene was found to be non-stereospecific considering the styrene geometry, indicating that a stepwise mechanism occurs in the reactions of this substrate. In this case, the relative endo/exo stereochemistry observed in the oxa-Povarov reactions was generally found to be opposite to that observed for the reactions with cyclopentene 7 and styrene 12.
Our theoretical studies on I-DA reactions involving iminium cations indicated that both one-step and stepwise mechanisms can be found. The I-DA reaction of iminium cation 1 has one-step mechanism.6 However, the presence of the strong electron-withdrawing piridinium substituent in iminium cation 4 enables the stabilization of a feasible intermediate once the first C–C single bond is completely formed, making the corresponding I-DA reaction stepwise.7 However, the very low activation energy associated with the ring-closure process in the stepwise reaction of iminium cation 4, 0.03 kcal mol−1,7 makes the characterization of the corresponding intermediates experimentally unfeasible, being the distinction between two-stage one-step and stepwise mechanisms non-relevant.
Herein, we report a DFT study of the mechanism of the I-DA reactions of cationic aryl oxonium 6 involved in the oxa-Povarov reactions and carried out experimentally by Batey et al.12 For this purpose, the I-DA reactions between cationic aryl oxonium 6 and cyclopentene 7, and styrene 12 will be analyzed (see Scheme 4). Our proposal is to perform a characterization of the molecular mechanisms involved in these I-DA reactions. An electron localization function (ELF)13 analysis of the bonding changes along one-step and stepwise reactions will be performed in order to establish the similarities between both mechanisms, and thus to propose a unified mechanism for bond-formation processes.
Computational methods
DFT computations were carried out using the B3LYP14 exchange-correlation functionals, together with the standard 6-31G(d) basis set.15 The optimizations were carried out using the Berny analytical gradient optimization method.16 The stationary points were characterized by frequency computations in order to verify that TSs have one and only one imaginary frequency. The IRC paths17 were traced in order to check the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.18 Solvent effects of dichloromethane (DCM) were taken into account through single point energy calculations using the polarisable continuum model (PCM) as developed by Tomasi's group19 in the framework of the self-consistent reaction field (SCRF).20 The 6-311G* basis set was used in the single point energy calculations in DCM. The electronic structures of stationary points were analyzed by the natural bond orbital (NBO) method21 and by the ELF topological analysis, η(r).13 The ELF study was performed with the TopMod program22 using the corresponding monodeterminantal wavefunctions of the selected structures of the IRC. All computations were carried out with the Gaussian 09 suite of programs.23The global electrophilicity index,24 ω, is given by the following expression, ω = (μ2/2η), in terms of the electronic chemical potential μ and the chemical hardness η. Both quantities may be approached in terms of the one-electron energies of the frontier molecular orbital HOMO and LUMO, εH and εL, as μ ≈ (εH + εL)/2 and η ≈ (εL − εH), respectively.25 Recently, we introduced an empirical (relative) nucleophilicity index,26 N, based on the HOMO energies obtained within the Kohn–Sham scheme,27 and defined as N = EHOMO(Nu) − EHOMO(TCE). The nucleophilicity is referred to tetracyanoethylene (TCE), because it presents the lowest HOMO energy in a large series of molecules already investigated in the context of polar cycloadditions. This choice allows us to handle conveniently a nucleophilicity scale of positive values. Radical Pok Parr functions,28 and P−k nucleophilic Parr functions,29 were obtained through the analysis of the Mulliken atomic spin density (ASD) of the neutral radical of the cations and the radical cation of the dienophiles.
Results and discussion
(i) Energetic, geometrical and electronic aspects of the oxa-Povarov reactions of the cationic aryl oxonium 6 with cyclopentene 7 and styrene 12
The oxa-Povarov reactions of the cationic aryl oxonium 6 with cyclopentene 7 and styrene 12 begin by an I-DA reaction to yield the corresponding formal [4+ + 2] CAs 8 and 13, which by a last proton abstraction afford chromans 9 and 14. The I-DA reaction between aryl oxonium 6 and cyclopentene 7 can take place along two stereoisomeric channels; the endo and the exo ones. Along the endo channel, the carbocyclic skeleton of cyclopentene 7 approaches over the O2 oxygen of aryl oxonium 6. An exploration of the potential energy surface (PES) associated with the endo and exo channels showed that this I-DA reaction takes place through a one-step mechanism (see Scheme 5). Thus, one TS and one formal [4+ + 2] CA were located and characterized for each one of the two stereoisomeric channels. Relative energies in DCM are summarized in Table 1. Energy discussion will be done using relative energies in DCM. Total energies are given in Table S1 in ESI.† | ||
| Scheme 5 | ||
| TS1n | 4.7 |
| TS1x | 3.8 |
| 8 | −21.0 |
| 10 | −24.0 |
In gas phase, both TSs are located below the reagents (see ESI†). However, when solvent effects of DCM are considered the activation energies become positive. As expected, this I-DA reaction presents very low activation energies: 4.7 (TS1n) and 3.8 (TS1x) kcal mol−1, the reaction being strongly exothermic. Both gas phase and in DCM calculations yield this I-DA reaction slightly exo selective. Conversion of formal [4+ + 2] CAs 8 and 10 into the final chromans 9-endo and 11-exo demands a proton abstraction by a basic species. When the acetate counterion was used as the basic species, the conversion of the cationic 8 and 10 into the final neutral chromans 9-endo and 11-exo took place without any activation barrier.
Due to the asymmetry of styrene 12, the I-DA reaction between aryl oxonium 6 and 12 can take place along four competitive channels: two regioisomeric, the meta and ortho channels, and two stereoisomeric channels, the endo and exo ones. Interestingly, an exploration of the PES associated with this I-DA reaction showed that the two regioisomeric channels present different mechanisms; the endo and exo stereoisomeric channels associated with the meta regioisomeric channels yield CAs 14-endo and 14-exo through a two-step mechanism, while those associated with the ortho channels present an one-step mechanism. Consequently, for the two endo and exo stereoisomeric meta channels two TSs, one intermediate, and one CA were found, while one TS and one CA were found along the ortho ones (see Scheme 6). Relative energies in DCM are summarized in Table 2. Total energies are given in Table S2 in ESI.†
 | ||
| Scheme 6 | ||
| TS21n | 0.6 | TS21x | −0.1 |
| IN1 | −9.8 | IN2 | −10.4 |
| TS22n | −8.8 | TS22x | −8.4 |
| 13 | −19.2 | 15 | −19.9 |
| TS3n | 14.0 | TS3x | 15.1 |
| 17 | −15.7 | 18 | −19.8 |
As in the I-DA reaction of oxonium cation 6 with cyclopentene 7, the TSs associated with the attack of the non-substituted C6 carbon of styrene 12 on the C1 carbon of 6 are located below the reagents. When solvent effects are considered, the relative energies of TSs are 0.6 and −0.1 kcal mol−1. Calculations also yield this I-DA reaction to be slightly exo selective. The reaction of oxonium cation 6 with styrene 12 is faster than that with cyclopentene 7 as a consequence of the more nucleophilic character of the former (see below). Formation of the corresponding cationic intermediates is exothermic by −9.8 (IN1) and −10.4 (IN2) kcal mol−1. However, with an unappreciable activation barrier below 2.0 kcal mol−1, they turn into the corresponding cationic CAs. Formation of the meta CAs are exothermic by −19.2 (13) and −19.9 (15) kcal mol−1.
Along the ortho channel, formation of formal [4+ + 2] CAs 17 and 18 through TS3n and TS3x present activation energies of 14.0 and 15.1 kcal mol−1, respectively. Consequently, the I-DA reaction between oxonium cation 6 with styrene 12 is completely regioselective.
As in the I-DA reaction of oxonium cation 6 with cyclopentene 7, when the acetate counterion was used as the basic species, the conversion of cationic 13 and 15 into the final neutral chromans 14-endo and 16-exo took place without any activation barrier.
It is noteworthy that although DFT calculations suggest that the I-DA reaction between oxonium cation 6 with styrene 12 takes place through a stepwise mechanism, the very low activation energy associated with the ring-closure process at the corresponding intermediates IN1 and IN2, makes the characterization of these stepwise mechanisms experimentally unfeasible.
The geometry of the TSs involved in the I-DA reactions between oxonium cation 6 and cyclopentene 7 and styrene 12 are given in Fig. 1 and 2. At the TSs involved the I-DA reaction between oxonium cation 6 and cyclopentene 7, the distances between the C1–C6 and C4–C5 carbon atoms are 2.006 and 3.003 Å at TS1n, and 2.020 and 3.201 Å at TS1x, respectively. The extent of the asynchronicity of bond-formation can be measured by means of the difference between the bond lengths of the two single bonds that are being formed in the reaction, i.e. Δd = d(C4–C5) – d(C1–C6). The values at the TSs are 1.00 at TS1n, and 1.18 at TS1x. These rather high values, which are similar to that found in the I-DA reaction between iminium cation 1 and Cp 2 (Δl = 1.18)6 (see Scheme 2), show the high asynchronicity in bond formation in I-DA reactions.
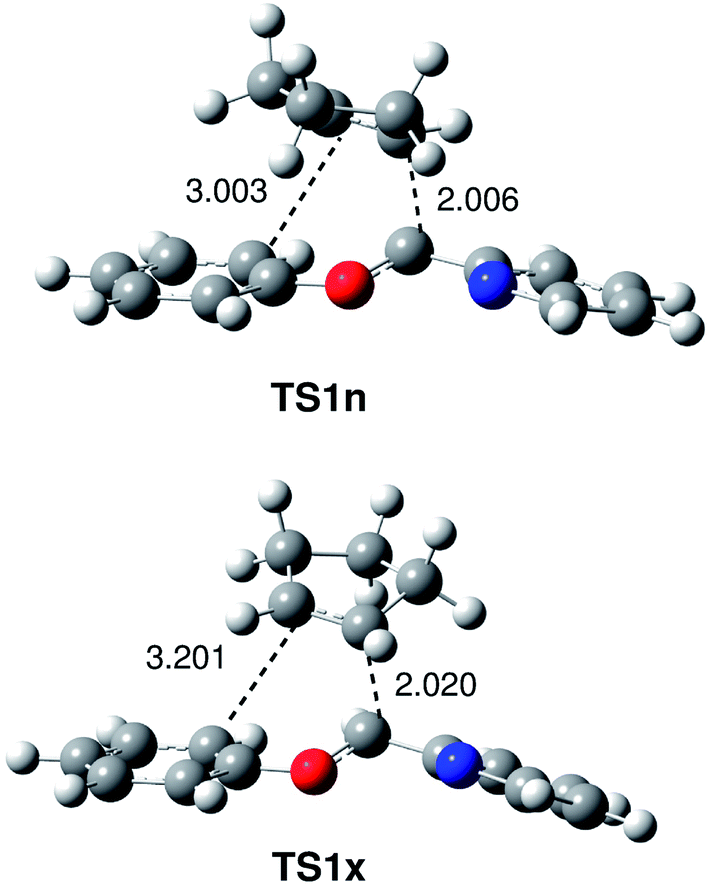 | ||
| Fig. 1 Transition state structures involved in the I-DA reactions between oxonium cation 6 and cyclopentene 7. Lengths are given in Angstroms. | ||
 | ||
| Fig. 2 Transition state structures involved in the I-DA reactions between oxonium cation 6 and styrene 12. Lengths are given in Angstroms. | ||
At the TSs and intermediates involved in the meta regioisomeric channels of the I-DA reaction between oxonium cation 6 and styrene 12, the distances between the C1 and C6, and the C4 and C5 carbons are 2.369 and 3.411 Å at TS21n, 2.314 and 3.769 Å at TS21x, 1.596 and 2.735 Å at IN1, 1.573 and 2.877 Å at IN2, 1.569 and 2.197 Å at TS22n, 1.559 and 2.248 Å at TS22x, respectively, while at the TSs involved in the ortho regioisomeric channels, the distances between the C1 and C5, and the C4 and C6 carbons are 1.916 and 2.524 Å at TS3n, and 1.937 and 2.630 Å at TS3x, respectively. The asynchronicity at the most favorable meta TSs are 1.04 at TS21n, and 1.47 at TS21x.
The most favorable meta TS21n and TS21x associated with the I-DA reaction between oxonium cation 6 and styrene 12 are earlier than TS1n and TS1x associated with the I-DA reaction with cyclopentene 7, in clear agreement with the lower activation energy found at the former.30 These stereoisomeric TSs, belonging to distinct one-step and stepwise mechanisms, present a high asynchronicity, Δl > 1; they are associated with two-center interactions resulting from the nucleophilic attack of cyclopentene 7 or styrene 12 on the C1 carbon of oxonium cation 6. It is noteworthy that in the four TSs, the distance between the C5 and C6 carbon atoms involved in the formation of the second C–C single bond is above of 3.0 Å, indicating no bonding interactions.
In order to establish the solvent effects on the geometry optimizations and relative energies, the stationary points involved in the I-DA reaction between oxonium cation 6 and cyclopentene 7 were fully optimized in DCM at the B3LYP/6-311G* level. The total and relative energies are given in Table S1† while the geometries of TS1n and TS1x in DCM are given in Fig. S1 in ESI.† Full optimizations in DCM at the B3LYP/6-311G* level did neither modify the relative energies (see Table S2†), nor the gas phase B3LYP/6-31G* geometries (see Fig. 1 and S1†). The IRCs from these TSs to reagents and product in DCM corroborated the two-stage one-step mechanism of this I-DA reaction.
Gas phase and implicit DCM solvation yield these I-DA reactions slightly exo selective, while experimentally being endo selective. Recently, we have established a relationship between the GCT in a P-DA reaction and the endo selectivity.31 In a P-DA reaction, the favourable electrostatic interactions that appear between the two opposite charged frameworks in the endo rearrangement are responsible for the endo selectivity. However, in I-DA reactions the two frameworks are charged with the same sign, i.e. these electrostatic interactions are unfavourable. Thus, weak interactions present in the endo TSs could be responsible for the endo stereoselectivity found in these I-DA reactions. Due to the weakness of the B3LYP functional to compute this type of interactions, single point energy calculations using the MPWB1K global-hybrid meta-GGA functional,32 which include long-range corrected and dispersion corrected methods, were performed at the stereoisomeric TSs TS1n and TS1x, and TS21n and TS21x. The MPWB1K/6-311G* energies in DCM are given in Table S3 in the ESI.† Calculations at the MPWB1K/6-311G* level in DCM render the two I-DA reactions slightly endo selective in agreement with the experimental results.33 Consequently, it appears that weak interactions present in the endo TSs are responsible for the endo stereoselectivity found in these I-DA reactions.
Finally, the GCT along these I-DA reactions was computed by two distinct methods: (i) by using the natural atomic charges obtained from a natural population analysis (NPA) at the TSs and intermediates associated with the endo stereoisomeric channels of I-DA reactions between oxonium cation 6 and cyclopentene 7, and styrene 12, and (ii) the integration of the electron-density of the ELF basins (see below). To compute the GCT at TSs and intermediates, the corresponding structures were divided between the oxonium cation framework and the dienophile one. The GCT is summarized in Table 3.
| d(C1–C6) | NPA | ELF | ||
|---|---|---|---|---|
| 6 + 7 | TS1n | 2.00 | 0.42 | 0.42 |
| 1.95 | 0.45 | 0.40 | ||
| 1.90 | 0.47 | 0.48 | ||
| P13 | 1.62 | 0.52 | 0.53 | |
| 6 + 12 | TS21n | 2.37 | 0.31 | 0.31 |
| 2.00 | 0.53 | 0.49 | ||
| 1.97 | 0.54 | 0.56 | ||
| IN1 | 1.53 | 0.67 | 0.65 | |
| d(C1–C5) | ||||
| TS3n | 1.92 | 0.36 | 0.36 | |
| 1.88 | 0.37 | 0.35 | ||
A comparison between the two methods used for the computation of the GCT indicates that there are no significant differences. The positive charge found at the cyclopentene or styrene frameworks at the TSs, between 0.42 e and 0.31 e, indicates that at these I-DA reactions a high amount of the electron density has been transferred from cyclopentene and styrene to the strong electrophilic oxonium cation 6. Note that while in P-DA reactions zwitterionic species are generated along the reactions, in I-DA reactions they remain cationic or anionic species. At the most favorable TS21n, the GCT, 0.31 e, is lower than that at TS1n, 0.42 e, as a consequence of the earlier character of the former. However, when the GCT is analyzed at one point of the IRC after passing TS21n with a C–C distance of 2.0 Å, similar to that at TS1n, the GCT becomes higher at the I-DA of styrene 12 as a consequence of the more nucleophilic character of styrene 12 than cyclopentene 7 (see below).
A comparison of the geometrical and electronic parameters of TS1n and TS21n indicates that both TSs show a great similarity, suggesting that the subsequent events in bonding changes along the reaction coordinates, i.e. participation of these TSs in one-step or two-step mechanisms, do not depend on the structure of these TSs.
(ii) ELF bonding analysis along the formation of the first C–C bond in I-DA reactions of oxonium cation 6 with cyclopentene 7 and styrene 12. Characterization of the C–C bond formation in ionic processes
Several theoretical studies have shown that the topological ELF analysis along a reaction path can be used as a valuable tool to understand the bonding changes along the reaction path.34 After an analysis of the electron density, the ELF provides basins, which are the domains in which the probability of finding an electron pair is maximal.13 The basins are classified as core basins and valence basins. The latter are characterized by the synaptic order, i.e., the number of atomic valence shells in which they participate. Thus, there are monosynaptic, disynaptic, trisynaptic basins and so on.35 Monosynaptic basins, labelled V(A), correspond to the lone pairs or non-bonding regions, while disynaptic basins connect the core of two nuclei A and B and, thus, correspond to a bonding region between A and B and are labelled V(A, B). This description recovers the Lewis bonding model, providing a very suggestive graphical representation of the molecular system.Recently, Domingo et al. have shown that the C–C single bond formation in both non-polar and polar organic reactions begins in the short C–C distance range of 1.9–2.0 Å by merging two monosynaptic basins, V(Cx) and V(Cy), into a new disynaptic basin V(Cx, Cy) associated with the formation of the new Cx–Cy single bond.36 The Cx and Cy carbons characterized by the presence of the monosynaptic basins, V(Cx) and V(Cy), have been called pseudoradical centers.37
In order to understand the C–C bond-formation process along these one-step and stepwise I-DA reactions, a topological ELF analysis of some relevant points of the corresponding IRCs was carried out. The most relevant ELF valence basins and their corresponding N populations of selected points along the reaction path are given in Table 4. On the other hand, the most relevant ELF attractors at some selected points of the IRCs of the one-step and two-step I-DA reactions of oxonium cation 6 with cyclopentene 7 and with styrene 12 are shown in Fig. 3.
| Cyclopentene 7 | Styrene 12 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| TS1n | P11 | P12 | P13 | P14 | TS21n | P21 | P22 | IN1 | TS22n | |
| d1(C1–C6) | 2.005 | 1.952 | 1.897 | 1.616 | 1.574 | 2.369 | 2.001 | 1.973 | 1.596 | 1.569 |
| d2(C4–C5) | 3.003 | 2.988 | 2.971 | 2.612 | 2.200 | 3.411 | 3.340 | 3.336 | 2.735 | 2.196 |
| V(C1) | 0.42 | 0.48 | — | — | — | — | 0.46 | — | — | — |
| V(C6) | 0.45 | 0.51 | — | — | — | — | 0.56 | — | — | — |
| V(C1, C6) | — | — | 1.11 | 1.70 | 1.84 | — | — | 1.08 | 1.76 | 1.86 |
| V(C4) | — | — | — | — | 0.13 | — | — | — | — | 0.31 |
| V(C5) | — | — | — | — | 0.44 | — | — | — | — | — |
| V(O2, C1) | 1.66 | 1.62 | 1.61 | 1.38 | 1.38 | 1.93 | 1.68 | 1.62 | 1.38 | 1.38 |
| V(C3, O2) | 1.55 | 1.57 | 1.54 | 1.56 | 1.66 | 1.61 | 1.55 | 1.54 | 1.58 | 1.67 |
| V(C3, C4) | 2.87 | 2.89 | 2.86 | 2.81 | 2.53 | 2.83 | 2.90 | 2.92 | 2.83 | 2.60 |
| V(C5, C6) | 2.69 | 2.61 | 2.55 | 2.24 | 2.15 | 3.14 | 2.53 | 2.48 | 2.16 | 2.11 |
 |
||||||||||
 | ||
| Fig. 3 Most relevant ELF attractors at some selected points of the IRCs of the one-step and two-step I-DA reactions of oxonium cation 6 with cyclopentene 7 and styrene 12. | ||
Along the endo reactive channel associated with the I-DA reactions of oxonium cation 6 with cyclopentene 7 and styrene 12, five selected structures were analyzed: (i) the TSs, (ii) the IRC structures in which the two monosynaptic basins associated with the two pseudoradical centers appear; (iii) the IRC structures associated with the formation of the first C1–C6 single bond; (iv) the structures in which the C1–C6 is completely formed; and (v) the structures associated with the formation of the second C4–C5 single bond.
Firstly, the two-step I-DA reaction between oxonium cation 6 and styrene 12 is analyzed. At TS21n, d1 = 2.37 Å and d2 = 3.41 Å, no monosynaptic basin appears at the C6 carbon of styrene 12 and the C1 carbon of oxonium cation 6. At this TS, the C5–C6 bonding region of styrene is characterized by the V(C5, C6) disynaptic basin, integrating 3.14 e. At P21, d1 = 2.00 Å and d2 = 3.34 Å, two monosynaptic basins, V(C1) and V(C6), integrating 0.46 e and 0.56 e, appear at the most nucleophilic C6 carbon of styrene 9, and the most electrophilic C1 carbon of oxonium cation 6. These monosynaptic basins are associated to the two pseudoradical centers37 responsible for the subsequent C1–C6 bond formation. At P22, d1 = 1.97 Å and d2 = 3.34 Å, the two V(C1) and V(C6) monosynaptic basins have merged into a new disynaptic basin, V(C1, C6), which integrates 1.08 e, indicating that the first C1–C6 single bond is already being formed. On going from P22 to intermediate IN1, the electron population of the V(C1, C6) disynaptic basin increases its electron density to reach 1.76 e at IN1, d1 = 1.60 Å and d2 = 2.74 Å. At this intermediate no monosynaptic basins appears at the C4 and C5 carbons indicating that, at this stationary point, the formation of the second C4–C5 sigma bond does not have begun. Finally, at TS22n, d1 = 1.57 Å and d2 = 2.20 Å, while the V(C1, C6) disynaptic basin has reached 1.86 e, one monosynaptic basin, V(C4), integrating 0.31 e appears at the C4 carbon.
On the other hand, at TS1n associated with the nucleophilic attack of cyclopentene 7 on the C1 carbon of oxonium cation 6, the most relevant features of its electronic structure are the presence of two monosynaptic basins, V(C1) and V(C6), integrating 0.42 e and 0.45 e, at the most electrophilic center of oxonium cation 6 and the most nucleophilic center of cyclopentene 7. At this TS, the C5–C6 bonding region of the cyclopentene framework is characterized by the V(C5, C6) disynaptic basin, integrating 2.69 e. Considering that this region of the cyclopentene 7 is characterized by the presence of two disynaptic basins, V(C5, C6) and V′(C5, C6), integrating 3.59 e, the loss of electron density at this bonding region, 0.90 e, can be associated with the formation of the V(C6) monosynaptic basin and the GCT involved in these I-DA reactions, 0.42 e (see Table 3). The electronic structure of TS1n, d1 = 2.00 Å and d2 = 3.00 Å, is very similar to that at P11, d1 = 1.95 Å and d2 = 2.99 Å. At this point of the IRC, the electron density of the two V(C1) and V(C6) monosynaptic basins increases to reach 0.48 e and 0.51 e. The electronic structures of TS1n and P11 are similar to that at P21 associated with the stepwise I-DA reaction between oxonium cation 6 and styrene 12. At P12, d1 = 1.90 Å and d2 = 2.97 Å, the two V(C1) and V(C6) monosynaptic basins have merged into a new disynaptic basin, V(C1–C6), which integrates 1.11 e. At this point of the IRC, the first C1–C6 is already formed.
Interestingly, P11 and P12, associated to the one-step mechanism of the I-DA reaction between oxonium cation 6 and cyclopentene 7, have similar electronic structures that P21 and P22, associated with the two-step I-DA reaction between oxonium cation 6 and styrene 12, indicating a similar pattern in the formation of the first C1–C6 single bond, not being dependent on the molecular mechanism (see Fig. 3).
On going from P12 to P13, d1 = 1.62 Å and d2 = 2.61 Å, the electron population of the V(C1, C6) disynaptic basin increases to reach 1.70 e at the P13 structure. At this point of the IRC, no monosynaptic basin appears at the C4 and C5 carbons, indicating that, at this point of the IRC, the formation of the second C4–C5 bond has not begun. Remarkably, P13 geometrically and electronically resembles IN1.
Finally, at P14, d1 = 1.57 Å and d2 = 2.20 Å, while the V(C1, C6) disynaptic basin has reached 1.84 e, two monosynaptic basins, V(C4) and V(C5), integrating 0.13 e and 0.44 e, appear at the C4 and C5 carbons. The resulting merge of these monosynaptic basins yields a new V(C4, C5) disynaptic basin responsible for the formation of the second C4–C5 bond.
The positions of the selected structures used in the ELF bonding analysis along the one-step I-DA reaction of oxonium cation 6 with cyclopentene 7 and the two-step I-DA reaction of oxonium cation 6 with styrene 12 are shown in Fig. 4. The current ELF bonding analysis for the formation of the C1–C6 and C4–C5 single bonds allows the establishment of the resemblance between the two-stage one-step and the two-step mechanisms of these I-DA reactions. Both mechanisms are non-concerted bond-formation processes. In the first part of the reactions only the C1–C6 single bond is being formed by the nucleophilic attack of the C6 carbon of cyclopentene 7 or styrene 12 on the C1 carbon of oxonium cation 6. Only when the C1–C6 bond formation is completed at the point P13 or at IN1, the formation of the second C4–C5 bond begins at the second part of the reaction. While Fig. 3 shows the electronic and geometrical similarity between the selected points of both mechanisms, Fig. 4 shows the similar position of these points along the IRC. The only difference between both mechanisms is the relative energy of P13 and IN1. The presence of the phenyl substituent in styrene 12 allows for the stabilization of the positive charge that is developing at C5 along the nucleophilic attack of styrene 12, allowing the localization of IN1 as a stationary point. This behavior explains the absence of the V(C5) monosynaptic basin at TS22n. However, the low activation energy associated with the ring-closure process, 1.0 kcal mol−1, makes it very difficult to experimentally characterize IN1, a behavior that makes both mechanisms kinetically very similar.
 | ||
| Fig. 4 Schematic representation of the energy profiles along the one-step (in red) and two-step (in blue) mechanisms of the I-DA reactions of oxonium cation 6 with cyclopentene 7 and styrene 12. The non-stationary selected points of the IRC used in the ELF analysis are represented by points. | ||
(iii) Analysis of the global reactivity indices of the reagents involved in I-DA reactions
The I-DA reactions were analyzed using the reactivity indices defined within the conceptual DFT.38 The global descriptors, named electronic chemical potential μ, chemical hardness η, global electrophilicity ω, and global nucleophilicity N indices for the series of reagents shown in Schemes 2–4 are given in Table 5.| μ | η | ω | N | |
|---|---|---|---|---|
| 4 | −14.60 | 4.67 | 22.79 | −7.81 |
| 6 | −9.06 | 3.06 | 13.42 | −1.47 |
| 1 | −11.15 | 7.38 | 8.42 | −5.72 |
| 12 | −3.43 | 5.20 | 1.13 | 3.09 |
| 2 | −3.01 | 5.49 | 0.83 | 3.36 |
| 7 | −2.72 | 7.24 | 0.51 | 2.78 |
Iminium cations 1 and 4, and oxonium cation 6 have very high electrophilicity ω values, 8.42, 22.79, and 13.42 eV, respectively, due to their cationic nature. These high electrophilicities are responsible for the very low even negative activation energies found in gas phase. On the other hand, Cp 2, cyclopentene 7 and styrene 12, have high nucleophilicity indices, N = 3.36, 2.78 and 3.09 eV, respectively. The higher nucleophilic character of styrene 12, when compared to than cyclopentene 7, accounts for the larger reactivity of the former towards oxonium cation 6.
It is interesting to remark that in spite of the high electrophilic character of cations 1, 4 and 6, the ionic classification of these DA reactions is not a consequence of the high electrophilicity of neither the reagent nor the high GCT at the TSs, but the ionic character of reagents, TSs and cycloadducts. Thus, a high electrophilic neutral molecule such tretracyanoethylene, ω = 5.96 eV, could participate in P-DA reactions with high polar character towards neutral dienes, but not in a I-DA reaction.
Along a polar reaction involving asymmetric reagents, the most favorable reactive channel is that involving the initial C–C two-center interaction between the most electrophilic and nucleophilic center of both reagents.2 Recently, we have proposed the electrophilic P+k and nucleophilic P−k Parr functions derived from the excess of spin electron density reached via a GCT process from the nucleophile to the electrophile.29,39
In our reactivity model for the C–C bond formation via an ionic mechanism, a cationic species such as oxonium cation 6 receives an amount of electron density from the neutral nucleophiles, cyclopentene 7 and styrene 12, that in the extreme case to receiving an amount equivalent to one electron, the cationic species turns into a neutral radical species. Similarly, when the nucleophile is an anionic species, it also turns into a neutral radical species after losing one electron. Thus, we suggest the use of the recently proposed radical Pok Parr function28 to analyze the local reactivity in positively or negatively charged species. Accordingly, the radical Pok Parr function of oxonium cation 6, and nucleophilic P−k Parr functions cyclopentene 7 and styrene 12 are given in Fig. 5.
 | ||
| Fig. 5 Map of the ASD of the radical 6˙ and radical Pok Parr function of oxonium cation 6, and maps of the ASD of the radical cations 7˙+ and 12˙+, and nucleophilic P−k Parr functions of cyclopentene 7 and styrene 12. | ||
Analysis of the radical Pok Parr functions of oxonium cation 6 indicates that the C1 carbon is the most electrophilic center of this cationic species, PoC1 = 0.61, in clear agreement with the asynchronicity found in all TSs associated with the I-DA reactions of oxonium cation 6.
On the other hand, while cyclopentene 7 shows a symmetric nucleophilic activation at the olefinic C5 and C6 carbon atoms, P−C5 = P−C6 = 0.40, the asymmetric styrene 12 shows the largest nucleophilic activation at C6 carbon, P−C6 = 0.43, while the C5 carbon atom is not nucleophilically activated, P−C5 = 0.00.
Consequently, the most favourable regioisomeric channel associated with the I-DA reaction between the asymmetric oxonium cation 6 and styrene 12 will be that associated with the initial formation of the C1–C6 single bond, in clear agreement with the observed total regioselectivity.
Conclusions
The mechanism of the oxa-Povarov reactions of cationic aryl oxonium 6 with cyclopentene 7 and styrene 12 to yield chromans 9 and 14, experimentally reported by Batey et al.,12 have been studied using DFT methods at the B3LYP/6-31G* in gas phase level for geometries and the B3LYP/6-311G* in DCM level for energies as reaction models of I-DA reactions. These oxa-Povarov reactions begin by the I-DA reaction of oxonium ion 6 with alkenes 7 and 12 to yield the formal [4+ + 2] CAs 8 and 13, which by a rapid loss of a proton yield chromans 9 and 14. The I-DA reactions have no appreciable activation barriers as a consequence of the strong electrophilic character of the cationic aryl oxonium 6. Unlike P-DA reactions, in which the polar character of the reaction is developed along the cycloaddition, in I-DA reactions, the cationic or anionic character of the species remains along the reactions.While the reaction with cyclopentene 7 takes place along a two-stage one-step mechanism, the presence of a phenyl substituent in styrene 12 able to stabilize a positive charge, makes the mechanism two-steps when stabilizing the corresponding intermediate. However, the very low activation energy associated with the ring closure makes the characterization of the corresponding intermediate experimentally unfeasible. The final proton abstraction has not appreciable activation barrier.
An ELF bonding analysis of selected points along the IRCs of the one-step mechanism of the I-DA reaction between cationic aryl oxonium 6 and cyclopentene 7, and the two-step mechanism of the I-DA reaction between 6 and styrene 12, allows the establishment of a great similarity in bond formation along the two mechanisms. Both one-step and two-step mechanisms are non-concerted processes.
As in P-DA reactions, the formation of the first C–C single bond begins in the short range of 1.95–1.90 Å, via a C-to-C pseudodiradical coupling of the most electrophilic and nucleophilic centers of cationic aryl oxonium 6, the C1 carbon, and cyclopentene 7 and styrene 12, the C6 carbon. The electron-density of the pseudoradical center generated at the C1 carbon of the cationic aryl oxonium 6, with a population of 0.46–048 e, comes mainly from the GCT that takes place along I-DA reactions, ca. 0.47 e. This interesting finding rejects the use of the arrows used in all text books to symbolize the changes in electron density in polar and ionic reactions.36b
Analysis of the global reactivity indices indicates that the high electrophilic character of the cationic reagents is responsible for the negative activation energies found in gas phase. However, this behavior is not responsible for the classification of I-DA reactions.
Based on our reactivity model of the GCT along polar and ionic reactions, herein, we suggest the use of the recently proposed radical Pok Parr functions to characterize the most electrophilic centers in cationic species and the most nucleophilic centers in anionic species. Thus, the analysis of the radical Pok Parr functions in cationic aryl oxonium 6 and the analysis of the nucleophilic P−k Parr functions in cyclopentene 7 and styrene 12 allows the anticipation of the most favorable two-center interaction along the formation of the first C–C single bond at the high asynchronous TSs, as well as the regioselectivity in I-DA reactions involving asymmetric reagents.
Acknowledgements
This work has been supported by Fondecyt grant (no. 1100278) and by the University of Valencia (project UV-INV-AE13-139082). Professor Domingo also thanks Fondecyt for continuous support through Cooperación Internacional.References
- (a) W. Carruthers, Some Modern Methods of Organic Synthesis, Cambridge University Press, Cambridge, 1978 Search PubMed; (b) W. Carruthers, Cycloaddition Reactions in Organic Synthesis, Pergamon, Oxford, 1990 Search PubMed.
- L. R. Domingo and J. A. Sáez, Org. Biomol. Chem., 2009, 7, 3576 CAS.
- I. Fleming, Molecular Orbitals and Organic Chemical Reactions, Wiley, 2009 Search PubMed.
- (a) P. G. Gassman and D. A. Singleton, J. Org. Chem., 1986, 51, 3075 CrossRef CAS; (b) P. G. Gassman and D. A. Singleton, J. Am. Chem. Soc., 1987, 109, 2182 CrossRef CAS.
- M. V. Basaveswara Rao, J. Satyanarayana, H. Ham and H. Junjappa, Tetrahedron Lett., 1995, 36, 3385 CrossRef.
- L. R. Domingo, J. Org. Chem., 2001, 66, 3211 CrossRef CAS.
- L. R. Domingo, M. Oliva and J. Andrés, J. Org. Chem., 2001, 66, 6151 CrossRef CAS.
- (a) L. R. Domingo, M. Oliva and J. Andrés, J. Mol. Struct.: THEOCHEM, 2001, 544, 79 CrossRef CAS; (b) V. Tamilmani, C. A. Daul and P. Venuvanalingam, Chem. Phys. Lett., 2005, 354 CrossRef CAS; (c) T. Kudoh, T. Mori, M. Shirahama, M. Yamada, T. Ishikawa, S. Saito and H. Kobayashi, J. Am. Chem. Soc., 2007, 129, 4939 CrossRef CAS; (d) V. Tamilmani, D. Senthilnathan and P. Venuvanalingam, J. Chem. Sci., 2008, 120, 225 CrossRef CAS; (e) R. Castillo and J. Andrés, Lett. Org. Chem., 2011, 8, 104 CrossRef CAS.
- H. Mayr, A. R. Ofial, J. Sauer and B. Schmied, Eur. J. Org. Chem., 2000, 2013 CrossRef CAS.
- C. Hedberg, P. Pinho, P. Roth and P. G. Andersson, J. Org. Chem., 2000, 65, 2810 CrossRef CAS.
- L. R. Domingo, J. A. Saéz, R. J. Zaragozá and M. Arnó, J. Org. Chem., 2008, 73, 8791 CrossRef CAS.
- R. R. Taylor and R. A. Batey, J. Org. Chem., 2013, 78, 1404 CrossRef CAS.
- (a) A. Savin, A. D. Becke, J. Flad, R. Nesper, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed., 1991, 30, 409 CrossRef; (b) B. Silvi and A. Savin, Nature, 1994, 371, 683 CrossRef CAS; (c) A. Savin, B. Silvi and F. Colonna, Can. J. Chem., 1996, 74, 1088 CrossRef CAS; (d) A. Savin, R. Nesper, S. Wengert and T. F. Fassler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1808 CrossRef CAS.
- (a) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS; (b) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- W. J. Hehre, L. Radom, P. v. R. Schleyer and J. A. Pople, Ab initio Molecular Orbital Theory, Wiley, New York, 1986 Search PubMed.
- (a) H. B. Schlegel, J. Comput. Chem., 1982, 2, 214 CrossRef; (b) H. B. Schlegel, in Modern Electronic Structure Theory, ed. D. R. Yarkony, World Scientific Publishing, Singapore, 1994 Search PubMed.
- K. Fukui, J. Phys. Chem., 1970, 74, 4161 CrossRef CAS.
- (a) C. González and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523 CrossRef; (b) C. González and H. B. Schlegel, J. Chem. Phys., 1991, 95, 5853 CrossRef.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS; (b) B. Y. Simkin and I. Sheikhet, Quantum Chemical and Statistical Theory of Solutions-A Computational Approach, Ellis Horwod, London, 1995 Search PubMed.
- (a) E. Cances, B. Mennucci and J. Tomasi, J. Chem. Phys., 1997, 107, 3032 CrossRef CAS; (b) M. Cossi, V. Barone, R. Cammi and J. Tomasi, Chem. Phys. Lett., 1996, 255, 327 CrossRef CAS; (c) V. Barone, M. Cossi and J. Tomasi, J. Comput. Chem., 1998, 19, 404 CrossRef CAS.
- (a) A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735 CrossRef CAS; (b) A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899 CrossRef CAS.
- S. Noury, X. Krokidis, F. Fuster and B. Silvi, Comput. Chem., 1999, 23, 597 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- R. G. Parr, L. von Szentpaly and S. Liu, J. Am. Chem. Soc., 1999, 121, 1922 CrossRef CAS.
- (a) R. G. Parr and R. G. Pearson, J. Am. Chem. Soc., 1983, 105, 7512 CrossRef CAS; (b) R. G. Parr and W. Yang, Density Functional Theory of Atoms and Molecules, Oxford University Press, New York, 1989 Search PubMed.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, J. Org. Chem., 2008, 73, 4615 CrossRef CAS PubMed; (b) L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2011, 9, 7168 RSC.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, 1133 CrossRef.
- L. R. Domingo and P. Pérez, Org. Biomol. Chem., 2013, 11, 4350 CAS.
- L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 1486 RSC.
- G. S. Hammond, J. Am. Chem. Soc., 1955, 77, 334 CrossRef CAS.
- L. R. Domingo, P. Pérez, M. J. Aurell and J. A. Sáez, Curr. Org. Chem., 2012, 16, 2343 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2004, 108, 6908 CrossRef CAS.
- Full gas phase optimisation of TS1n and TS1x at the MPWB1K/6-31G* computational level yields the I-DA reaction between oxonium cation 6 and cyclopentene 7 slightly exo selective, ΔΔE(TS1n−TS1x) = −0.29 kcal mol−1. Consequently, the only change of the B3LYP functional by the MPW1K one is not sufficient to model the endo selectivity experimentally observed. These energy results indicate that the stereoselectivity in I-DA reactions is dependent on the computational level. Note that in P-DA reactions, any computational level furnishes the experimental endo selectivity.
- V. Polo, J. Andres, S. Berski, L. R. Domingo and B. Silvi, J. Phys. Chem. A, 2008, 112, 7128 CrossRef CAS PubMed.
- B. Silvi, J. Mol. Struct., 2002, 614, 3 CrossRef CAS.
- (a) L. R. Domingo, M. J. Aurell, P. Pérez and J. A. Sáez, RSC Adv., 2012, 2, 1334 RSC; (b) L. R. Domingo, P. Pérez and J. A. Sáez, Tetrahedron, 2013, 69, 107 CrossRef CAS; (c) L. R. Domingo, P. Pérez and J. A. Sáez, RSC Adv., 2013, 3, 7520 RSC.
- (a) L. R. Domingo, E. Chamorro and P. Pérez, Lett. Org. Chem., 2010, 432 CrossRef CAS; (b) L. R. Domingo and J. A. Saez, J. Org. Chem., 2011, 76, 373 CrossRef CAS PubMed.
- (a) P. Geerlings, F. De Proft and W. Langenaeker, Chem. Rev., 2003, 103, 1793 CrossRef CAS PubMed; (b) D. H. Ess, G. O. Jones and K. N. Houk, Adv. Synth. Catal., 2006, 348, 2337 CrossRef CAS.
- Our reactivity model based on the GCT process was proposed for the first time in 1942 by R. B. Woodward in the manuscript entitled “The mechanism of the Diels–Alder reaction”,40 and more recently by R. P. Johnson.41.
- R. W. Woodward, J. Am. Chem. Soc., 1942, 64, 3058 CrossRef CAS.
- K. J. Cahill and R. P. Johnson, J. Org. Chem., 2013, 78, 6809 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Total and relative energies in gas phase and DCM of the stationary points involved in the I-DA reaction of oxonium cation 6 with cyclopentene 7, and with styrene 12. MPWB1K/6-311G* energies in DCM of the stereoisomeric TSs involved in the I-DA reaction of oxonium cation 6 with cyclopentene 7 and styrene 12. B3LYP/6-311G* full optimized geometries of TS1n and TS1x in DCM. B3LYP/6-31G* computed total energies, unique frequency imaginary, and cartesian coordinates of the stationary points involved in reaction of cationic aryl oxonium 6 with cyclopentene 7, and with styrene 12. See DOI: 10.1039/c3ra47805j |
| This journal is © The Royal Society of Chemistry 2014 |