Taurospongins B and C, new acetylenic fatty acid derivatives possessing a taurine amide residue from a marine sponge of the family Spongiidae†
Takaaki Kubotaa,
Haruna Suzukia,
Azusa Takahashi-Nakaguchib,
Jane Fromontc,
Tohru Gonoib and
Jun'ichi Kobayashi*a
aGraduate School of Pharmaceutical Sciences, Hokkaido University, Sapporo 060-0812, Japan. E-mail: jkobay@pharm.hokudai.ac.jp; Fax: +81 11 706 4989; Tel: +81 11 706 3239
bMedical Mycology Research Center, Chiba University, Chiba 260-0856, Japan. E-mail: gonoi@faculty.chiba-u.jp; Fax: +81 43 226 2486; Tel: +81 43 226 2492
cWestern Australian Museum, Locked Bag 49, Welshpool DC, WA 6986, Australia. E-mail: Jane.Fromont@museum.wa.gov.au; Fax: +61 8 9212 3882; Tel: +61 8 9212 3745
First published on 7th February 2014
Abstract
Two new acetylenic fatty acid derivatives possessing a taurine amide residue, taurospongins B (1) and C (2), have been isolated from an Okinawan marine sponge of the family Spongiidae. The gross structures of 1 and 2 were elucidated on the basis of their spectral data, especially 2D NMR and FABMS/MS data. The absolute configurations for 1 and 2 were established by chemical means. Taurospongin C (2) showed inhibitory activity against Cryptococcus neoformans.
Introduction
Marine sponges of the family Spongiidae have been demonstrated to be a rich source of unique bioactive meroterpenoids1,2 and acetylenic fatty acid derivatives.3,4 During our search for bioactive metabolites from marine organisms, we investigated the extract of a sponge family Spongiidae (SS-1202), which resulted in the isolation of two new acetylenic fatty acid derivatives possessing a taurine amide residue, taurospongins B (1) and C (2). Here we describe the isolation and structure elucidation of 1 and 2 (Fig. 1). | ||
| Fig. 1 Taurospongins B (1), C (2), and A (3). | ||
Results and discussion
The sponge family Spongiidae collected at Okinawa, was extracted with MeOH. After evaporation, the MeOH extract was partitioned stepwise between organic solvents (EtOAc and n-BuOH) and H2O. A part of n-BuOH soluble materials was fractionated by gel filtration (Sephadex LH-20, MeOH). A fraction eluted in a relatively early stage was purified by C18 column chromatography (MeOH–H2O) and SiO2 column chromatography (CH3Cl–MeOH) to afford taurospongins B (1, 1.7 mg, 0.00048%, wet weight) and C (2, 4.2 mg, 0.0012%).Taurospongin B (1) was obtained as an optically active colorless amorphous solid. The molecular formula of 1 was revealed to be C38H69NO7S by HRESIMS data [m/z 682.47036 (M − H)−, Δ −1.84 mmu]. IR absorptions indicated the existence of hydroxy (3421 cm−1), ester carbonyl (1732 cm−1), and amide carbonyl (1646 cm−1) functionalities. The inspection of the HMQC and HMBC spectra with 1H and 13C NMR data disclosed that 1 consists of ester and amide carbonyls, a triple bond, a double bond, three methyls, twenty seven methylenes, an oxymethine, and an oxygenated quaternary carbon (Table 1). Analysis of the 1H–1H COSY and TOCSY spectra of 1 revealed connectivities of C-4 to C-10, C-2′ to C-3′, C-6′ to C-10′, C-23′ to C-25′, and C-1′′ to C-2′′. The geometry of a double bond between C-8′ and C-9′ was assigned as Z by the vicinal coupling constant (3JH-8′/H-9′ = 10.3 Hz). HMBC correlations of H2-1′′/C-1 and H2-2/C-1 clarified that N-bearing carbon C-1′′ (δC 37.3) and a carbonyl-bearing carbon C-2 (δC 48.6) were connected via an amide bond containing a carbonyl carbon C-1 (δC 174.6). Linkings of C-2, a methylene carbon C-4, and a methyl carbon C-11 through an oxygenated quaternary carbon C-3 (δC 73.4) were inferred from HMBC correlations of H3-11/C-2, H2-4/C-3, and H3-11/C-4. An HMBC correlation of H2-2′/C-1′ revealed that a carbonyl-bearing carbon C-2′ (δC 36.2) was attached to a carbonyl carbon C-1′ (δC 174.6). Linkage of C-3′ and C-6′ by a triple bond between acetylenic carbons C-4′ (δC 80.2) and C-5′ (δC 82.2) was uncovered by HMBC correlations of H2-3′/C-4′ and H2-6′/C-5′. The chemical shift of a proton H-7 (δC 4.98) of an oxygenated carbon C-7 (δC 76.2) implied that C-7 was esterified to C-1′. These data and the molecular formula of 1 indicated an attachment of a sulfo group to C-2′′ and the connection of C-10′ and C-23′ by a methylene chain (Fig. 2).
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δHa multi (J in Hz) | δCb | δHa multi (J in Hz) | δCb | |
| a Recorded at 600 MHz.b Recorded at 150 MHz.c 2H.d 3H.e 24H.f 12C.g J-values were not determined because of overlapping with other signals.h These signals might be exchange.i These signals might be exchange.j These signals might be exchange.k These signals might be exchange.l These signals might be exchange. | ||||
| 1 | — | 174.6 | — | 174.5k |
| 2 | 2.40 d (14.0) | 48.6 | 2.36c m | 48.4 |
| 2.34 d (14.0) | ||||
| 3 | — | 73.4 | — | 74.2 |
| 4 | 1.59 mg | 43.8 | 1.59 mg | 43.9 |
| 1.53 mg | 1.52 mg | |||
| 5 | 1.44c mg | 21.6 | 1.47c mg | 21.8 |
| 6 | 1.61c mg | 36.6 | 1.64c mg | 36.7 |
| 7 | 4.98 m | 76.2 | 3.67 mg | 70.1 |
| 8 | 1.59c mg | 38.3 | 1.85 mg | 45.3 |
| 1.64 mg | ||||
| 9 | 1.43c mg | 20.5h | 5.15 m | 71.3 |
| 10 | 0.96d t (7.4) | 15.1i | 1.30d d (6.3) | 20.7 |
| 11 | 1.25d s | 27.6 | 1.27d s | 27.7 |
| 1′ | — | 174.6 | — | 174.3k |
| 2′ | 2.51c m | 36.2 | 2.50c m | 36.2 |
| 3′ | 2.48c m | 16.5 | 2.48c m | 16.4 |
| 4′ | — | 80.2 | — | 80.1 |
| 5′ | — | 82.2 | — | 82.2 |
| 6′ | 2.18c m | 20.7h | 2.18c m | 20.7 |
| 7′ | 2.23c m | 28.8j | 2.23c m | 28.7l |
| 8′ | 5.44 dt (10.3, 5.6) | 130.0 | 5.44 dt (10.7, 5.4) | 130.0 |
| 9′ | 5.45 dt (10.3, 5.6) | 132.8 | 5.46 dt (10.7, 5.4) | 132.8 |
| 10′ | 2.09c m | 29.0j | 2.09c q (6.4) | 29.0l |
| 11′–22′ | 1.52–1.15e mg | 31.7–31.1f | 1.52–1.15e m | 31.7–31.1f |
| 23′ | 1.33c mg | 33.9 | 1.38c m | 33.9 |
| 24′ | 1.38c mg | 24.5 | 1.33c m | 24.5 |
| 25′ | 0.94 t (7.1) | 15.2i | 0.94 t (7.0) | 15.3 |
| 1′′ | 3.66 t (6.5) | 37.3 | 3.67 mg | 37.3 |
| 2′′ | 3.02 t (6.5) | 52.2 | 3.03 t (6.6) | 52.2 |
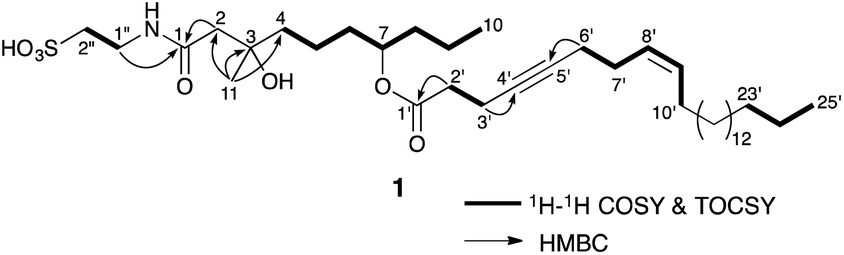 | ||
| Fig. 2 Selected 2D NMR correlations for taurospongins B (1). | ||
The structure of 1 elucidated from the NMR data was also confirmed by a charge-remote fragmentation pattern induced by a sulfo group observed in the FABMS/MS spectrum of 1 (Fig. 3). Thus, the gross structure of taurospongin B (1) was elucidated as shown.
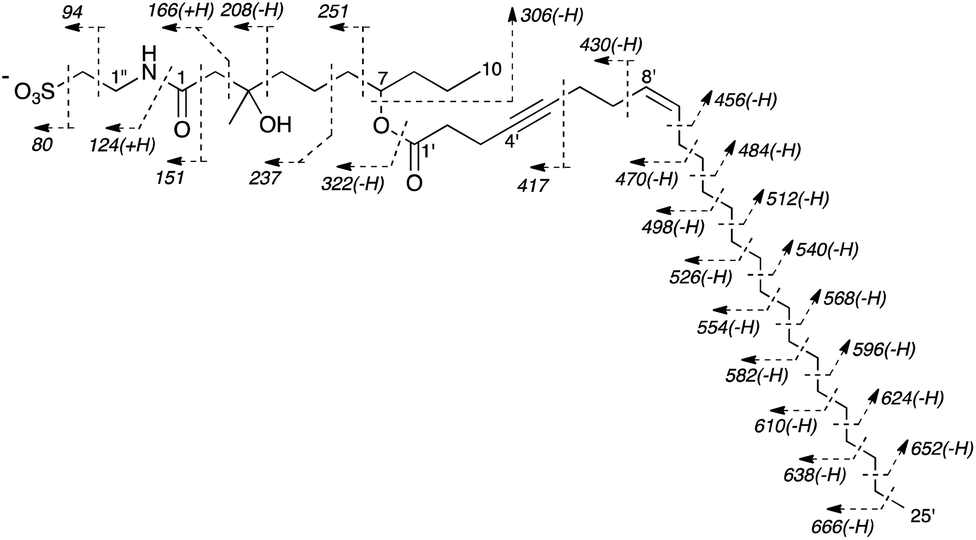 | ||
| Fig. 3 Fragmentation patterns observed in FABMS/MS spectrum of taurospongin B (1) [precursor ion, m/z 682 (M − H)−]. The m/z values were indicated in italics. | ||
The relative stereochemistry of taurospongin B (1) was established by comparison of the NMR data of taurineamide part (4) of 1 with its two possible diastereomers (6a and 6b). The taurineamide part (4) was obtained by methanolysis of an ester linkage of 1 with an acetylenic fatty acid part (5) of 1 (Scheme 1).4
 | ||
| Scheme 1 Methanolysis of taurospongin B (1). | ||
The taurine amides 6a and 6b were synthesized from D-mevalonolactone as follows (Schemes 2 and 3). A hydroxy group of D-mevalonolactone was protected by a TES group and then treated with DIBAL to give lactol (8), which was coupled with phosphorane by Wittig reaction. After protection of hydroxy group by a BOM group, the resulting chloro-olefin (10) was subjected to a coupling reaction with n-butylaldehyde to afford a mixture of acetylenic alcohols (11a and 11b), which was converted into an acetylenic ketone (12) by oxidation using AZADO.5
 | ||
| Scheme 2 Synthesis of 6a and 6b (part 1). | ||
 | ||
| Scheme 3 Synthesis of 6a and 6b (part 2). | ||
The acetylenic ketone (12) was derived to chiral acetylenic alcohol (11a) by a Noyori asymmetric transfer hydrogenation using Ru[(S,S)-Tsdpen](p-cymene).6 The absolute stereochemistry at C-7 of 11a was verified by modified Mosher's method7 to be R-configuration. After the hydroxy group of the 11a was protected by TBS group, deprotection of BOM group and reduction of the triple bond were accomplished by using Pearlman's catalyst.8 The resulting primary alcohol (14a) was oxidized to carboxylic acid (15a) by combination of Parikh–Doering oxidation9 and Lindgren–Kraus oxidation.10 Finally, the carboxylic acid was coupled with taurine by amidation using DMT-MM11 to obtain 3S,7R-isomer (6a). The 3S,7S-isomer (6b) was prepared from the acetylenic ketone (12) in a similar manner using Ru[(R,R)-Tsdpen](p-cymene).6 Since the 1H NMR spectra of taurineamide part (4) of taurospongin B (1) was coincident with that of 6a, relative relasionship for two hydroxy groups of 4 were uncoverd to be syn relationship.
The absolute configuration at C-7 of taurospongin B (1) was established by modified Mosher's method.7 Treatment of taurineamide part (4) with (R)-(−)- and (S)-(+)-MTPACl gave (S)- and (R)-MTPA esters of 4, respectively. Δδ values obtained from 1H NMR data of MTPA esters of 4 (Fig. 4) indicated that 4 was an enantiomer of 6a and the absolute configuration at C-7 of 4 was R. Therefore, the absolute configurations at C-3 and C-7 of taurospongin B (1) were assigned as both R.
 | ||
| Fig. 4 Δδ values [Δδ (in ppm) = δS − δR] obtained for the (S)- and (R)-MTPA esters of taurineamide part (4) of taurospongin B (1). Δδ values were indicated in italic. | ||
Taurospongin C (2) was obtained as an optically active colorless amorphous solid. The molecular formula of 2 was elucidated as C38H69NO8S by HRESIMS data [m/z 698.46739 (M − H)−, Δ +0.28 mmu]. IR absorptions suggested that 2 possesses hydroxy (3369 cm−1), ester carbonyl (1730 cm−1), and amide carbonyl (1636 cm−1) functionalitiess. The 1H and 13C NMR data of 2 were almost superimposable to those of taurospongin A (3),4,12,13 except for disappearance of signals derived from an acetyl group. In addition, the difference of the molecular formula between 2 and 3 implied that 2 was a 9-O-desacetyl form of 3. To verify the prediction, 2 was derived to 9-O-acetyl form by treatment of 2 with acetic anhydride and pyridine. Since the spectral data of 9-O-acetyl form of 2 was identical with those of 3, 2 was assigned as 9-O-desacetyl taurospongin A (3).
Antimicrobial assay14 of taurospongins B (1), C (2), and A (3) revealed that 2 and 3 exhibited mild and good antifungal activity, respectively, against Cryptococcus neoformans (MIC, 32 and 1 μg mL−1), while 1 did not show such activity (MIC > 32.0 μg mL−1). Taurospongins B (1), C (2), and A (3) were not active against other fungi Aspergillis niger, Trichophyton mentagrophytes, and Candida albicans (MIC > 32 μg mL−1), and bacteria Escherichia coli, Staphylococcus aureus, Bacillus subtilis, and Micrococcus luteus (IC50 > 32 μg mL−1). Taurospongins B (1), C (2), and A (3) did not show cytotoxicity (IC50 > 10 μg mL−1) against murine lymphoma L1210 and human epidermoid carcinoma KB cells in vitro.
Conclusions
Taurospongins B (1) and C (2), two new acetylenic fatty acid derivatives possessing a taurine amide residue, have been isolated from an Okinawan marine sponge of the family Spongiidae. The absolute structures of 1 and 2 were established by combination of spectroscopic analysis and synthetic chemistry. Taurospongins C (2) and A (3) showed mild and good antifungal activity, respectively, against Cryptococcus neoformans (MIC, 32 and 1 μg mL−1).Experimental section
General experimental procedures
Optical rotations were recorded on a JASCO P-1030 polarimeter. IR spectra were recorded on JASCO FT/IR-230 spectrometer. 1H and 13C NMR spectra were recorded on JEOL ECA400 and JEOL ECA500 spectrometers using 5 mm cell and a Bruker AMX-600 spectrometer using 2.5 mm micro cell. The 7.20 and 77.0 ppm resonances of residual CHCl3 and CDCl3, and 3.35 and 49.8 ppm resonances of residual CD2HOD and CD3OD were used as internal references for 1H and 13C NMR spectra, respectively. EIMS spectra were recorded on a JEOL JMS-700TZ mass spectrometer. ESIMS spectra were recorded on JEOL JMS-700TZ and Thermo Scientific Exactive mass spectrometers.Sponge description
The sponge (SS-1202, family Spongiidae) was collected at Unten Port, Okinawa, and kept frozen until used. The sponge was medium brown mound with a spiky, conulose surface, hispid with a fine adherent membrane, and alcohol brown stains. Dermis was unarmored. The sponge has a reticulate fibre skeleton with some pithing of fibres centrally. The reticulation is irregular and all fibres are uncored. Primary fibres are ∼50 μm wide. Secondary fibres are ∼40 μm wide with finer fibres between as a tertiary skeleton, 10 μm wide. The voucher specimen was deposited at Graduate School of Pharmaceutical Sciences, Hokkaido University.Extraction and isolation
The sponge of the family Spongiidae (SS-1202, 1.25 kg, wet weight) collected at Okinawa, was extracted with MeOH (2L × 2) to afford the extract (73.3 g), which was partitioned stepwise between organic solvents [EtOAc (500 mL × 3) and n-BuOH (500 mL × 3)] and H2O (500 mL) to give EtOAc-soluble materials (11.8 g) and n-BuOH-soluble materials (3.6 g). A part (1.0 g) of n-BuOH soluble materials was fractionated by gel filtration (Sephadex LH-20, GE Healthcare; eluent, MeOH). A fraction was purified by C18 column chromatography (Cosmosil 140 C18 PREP, Nakarai Tesque Inc.; eluent, MeOH–H2O, 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 to 100:0) and SiO2 column chromatography (Wakosil C-300, Wako Pure Chemical Industries, Ltd.; eluent, CH3Cl–MeOH, 95:5 to 0:100) to afford taurospongins B (1, 1.7 mg, 0.00048%, wet weight) and C (2, 4.2 mg, 0.0012%).
:1, 500 μl). After stirring for 12 h at 80 °C, the mixture was concentrated by Ar blowing. The residue was extracted with CHCl3, which was concentrated by Ar blowing to afford acetylenic fatty acid part (5, 0.5 mg, 1.28 μmol, 88%). The remaining CHCl3 insoluble material was taurineamide part (4, 0.4 mg, 1.23 μmol, 84%).:0 to 85:15) to afford 7 (1.81 g, 7.41 mmol, 90%); yellow oil; [α]21D − 28 (c 1.6, CHCl3); IR (neat) νmax 2957, 2877, 1738, 1458, 1223, 1005, 725 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.58 (m, 1H), 4.34 (m, 1H), 2.67 (brd, J = 17.2 Hz, 1H), 2.43 (d, J = 17.2 Hz, 1H), 1.84 (m, 2H), 1.39 (s, 3H), 0.94 (t, J = 8.0 Hz, 9H), 0.60 (q, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 170.2, 70.4, 66.1, 45.6, 36.9, 29.0, 6.8, 6.3; HRESIMS (pos.) m/z 267.13881 [calcd for C12H24O3NaSi (M + Na)+, Δ +0.12 mmu].:0 to 85:15) to afford 8 (1.81 g, 7.35 mmol, 97%), which was directly used in the next step; yellow oil; HRESIMS (pos.) m/z 269.15447 [calcd for C12H26O3NaSi (M + Na)+, Δ +0.13 mmu].:0 to 90:10) to afford 9 (1.29 g, 4.63 mmol, 62%), which was directly used in the next step; pale yellow oil; HRESIMS (pos.) m/z 301.13601 [calcd for C13H27O2ClNaSi (M + Na) +, Δ −0.10 mmu].:0 to 90:10) to afford 10 (1.56 g, 3.91 mmol, 84%), which was directly used in the next step; pale yellow oil; HRESIMS (pos.) m/z 421.19366 [calcd for C21H35O3ClNaSi (M + Na)+, Δ +0.04 mmu].:0 to 90:10) to afford mixture of 11a and 11b (1.21 g, 2.78 mmol, 79%), which was directly used in the next step; pale yellow oil; HRESIMS (pos.) m/z 457.27449 [calcd for C25H42O4NaSi (M + Na)+, Δ +0.03 mmu].:1, 30 mL) were added to the mixture, which was extracted with CH2Cl2. The organic layer was washed with brine, dried with MgSO4, and concentrated in vacuo. The residue was purified by a SiO2 column (n-hexane–EtOAc, 100:0 to 90:10) to afford 12 (1.16 g, 2.68 mmol, 95%); pale yellow oil; [α]22D + 5.0 (c 1.25, CHCl3); IR (neat) νmax 3019, 2876, 2211, 1673, 1455, 1377, 1240, 1171, 1114, 1045, 742, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 4.75 (s, 2H), 4.60 (s, 2H), 3.72 (m, 2H), 2.61 (d, J = 17.1 Hz, 1H), 2.54 (d, J = 17.1 Hz, 1H), 2.50 (t, J = 7.3 Hz, 1H), 1.96 (ddd, J = 13.9, 7.3, 6.8 Hz, 1H), 1.89 (ddd, J = 13.9, 7.5, 6.4 Hz, 1H), 1.69 (qt, J = 7.3, 7.3 Hz, 2H), 1.37 (s, 3H), 0.95 (qt, J = 7.8 Hz, 9H), 0.93 (t, J = 7.3 Hz, 3H), 0.60 (q, J = 7.8 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 188.1, 138.0, 128.4, 127.8, 127.7, 94.7, 91.1, 82.6, 73.9, 69.4, 64.1, 47.4, 41.6, 33.8, 28.1, 17.6, 13.5, 7.0, 6.7; HRESIMS (pos.) m/z 455.25877 [calcd for C25H40O4NaSi (M + Na)+, Δ −0.04 mmu].:0 to 80:20) to afford 11a (563 mg, 1.30 mmol, 97%, 97:3 dr); pale brown oil; [α]23D + 4.8 (c 1.23, CHCl3); IR (neat) νmax 3441, 2956, 2875, 1455, 1376, 1150, 1111, 1041, 733 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.76 (s, 2H), 4.61 (s, 2H), 4.33 (brt, J = 6.5 Hz, 1H), 3.74 (t, J = 7.4 Hz, 2H), 2.44 (dd, J = 16.4 and 1.5 Hz, 1H), 2.36 (dd, J = 16.4 and 1.8 Hz, 1H), 2.03–1.75 (m, 3H), 1.63 (m, 2H), 1.46 (m, 2H), 1.33 (s, 3H), 0.94 (t, J = 7.9 Hz, 9H), 0.93 (t, J = 7.3 Hz, 3H), 0.59 (q, J = 7.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 137.8, 128.4, 127.9, 127.7, 94.5, 83.4, 82.3, 74.0, 69.3, 64.4, 62.4, 41.2, 40.1, 33.4, 28.0, 18.4, 13.7, 7.0, 6.7; HRESIMS (pos.) m/z 457.27512 [calcd for C25H42O4NaSi (M + Na)+, Δ +0.66 mmu].:0 to 2:1) to afford (S)-MTPA ester of 11a (0.7 mg, 1.08 mmol, 94%); 1H NMR (600 MHz, CDCl3) δ 7.71–7.29 (m, 10H), 5.56 (t, J = 6.7 Hz, 1H), 4.73 (s, 2H), 4.59 (s, 2H), 3.71 (t, J = 7.3 Hz, 2H), 3.58 (s, 3H), 2.46 (d, J = 16.4 Hz, 1H), 2.38 (d, J = 16.4 Hz, 1H), 1.93 (m, 1H), 1.86 (m, 1H), 1.73 (m, 2H), 1.35 (m, 2H), 1.31 (s, 3H), 0.93 (t, J = 7.9 Hz, 9H), 0.87 (t, J = 7.3 Hz, 3H), 0.57 (q, J = 7.9 Hz, 6H); HRESIMS (pos.) m/z 673.31519 [calcd for C35H49O6F3NaSi (M + Na)+, Δ +0.92 mmu].:3 dr) by using [(1R,2R)-N-(p-toluenesulfonyl)-1,2-duphenylethanediamine]-(p-cymene)ruthenium(II) through the same procedure as described for preparation of 11a; pale brown oil; [α]23D + 8.4 (c 1.77, CHCl3); IR (neat) νmax 3447, 2956, 2875, 1455, 1376, 1150, 1112, 1041, 741 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.76 (s, 2H), 4.61 (s, 2H), 4.33 (brs, 1H), 3.74 (t, J = 7.3 Hz, 2H), 2.44 (dd, J = 16.4 and 1.8 Hz, 1H), 2.36 (dd, J = 16.4 and 1.8 Hz, 1H), 1.98 (m, 1H), 1.87 (m, 2H), 1.63 (m, 2H), 1.46 (m, 2H), 1.33 (s, 3H), 0.94 (t, J = 7.8 Hz, 9H), 0.93 (t, J = 7.4 Hz, 3H), 0.59 (q, J = 7.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 137.8, 128.4, 127.9, 127.7, 94.5, 83.4, 82.3, 74.0, 69.2, 64.4, 62.4, 41.2, 40.1, 33.4, 28.0, 18.5, 13.7, 7.1 6.6; HRESIMS (pos.) m/z 457.27408 [calcd for C25H42O4NaSi (M + Na)+, Δ −0.38 mmu].:0 to 90:10) to afford 13a (675 mg, 1.23 mmol, 95%); colorless oil; [α]24D − 14.6 (c 1.17, CHCl3); IR (neat) νmax 2956, 2876, 1456, 1251, 1111, 1041, 836, 776, 731 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.75 (s, 2H), 4.60 (s, 2H), 4.33 (brt, J = 6.3, 1H), 3.73 (t, J = 7.4 Hz, 2H), 2.43 (dd, J = 16.4 and 1.5 Hz, 1H), 2.34 (dd, J = 16.4 and 1.9 Hz, 1H), 1.98 (m, 1H), 1.86 (m, 1H), 1.61 (m, 2H), 1.42 (m, 2H), 1.33 (s, 3H), 0.94 (t, J = 7.9 Hz, 9H), 0.91 (t, J = 7.3 Hz, 3H), 0.90 (s, 9H), 0.58 (q, J = 7.9 Hz, 6H), 0.11 (s, 3H), 0.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 137.9, 128.4, 127.9, 127.6, 94.6, 84.0, 81.1, 74.1, 69.2, 64.4, 62.9, 41.1, 41.1, 33.5, 28.0, 25.8, 18.6, 18.2, 13.8, 7.1, 6.7, −4.5, −5.1; HRESIMS (pos.) m/z 571.36206 [calcd for C31H56O4NaSi2 (M + Na)+, Δ +1.13 mmu].:0 to 90:10) to afford 15a (2.0 mg, 0.0092 mmol, 77%); colorless oil; [α]18D − 5.9 (c 2.02, CHCl3); IR (neat) νmax 3410, 2923, 2853, 1717, 1562, 1456, 1240, 1129, 774 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.05 (brs, 1H), 3.64 (m, 1H), 2.56 (d, J = 15.6 Hz, 1H), 2.47 (d, J = 15.6 Hz, 1H), 1.61–1.29 (m, 10H), 1.28 (s, 3H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.8, 71.5, 71.4, 44.6, 41.6, 39.6, 37.2, 26.7, 19.9, 18.8, 14.1; HRESIMS (pos.) m/z 241.14118 [calcd for C11H22O4Na (M + Na)+, Δ +0.15 mmu].:0 to 50:50) to afford 6a (19.0 mg, 0.058 mmol, 53%); colorless oil; [α]20D + 3.5 (c 1.14, MeOH); IR (neat) νmax 3366, 2955, 2871, 2280, 1638, 1555, 1284, 1144, 1059, 938 cm−1; 1H NMR (600 MHz, CD3OD) δ 3.67 (brt, J = 6.7 Hz, 2H), 3.58 (m, 1H), 3.03 (t, J = 6.6 Hz, 2H), 2.43 (d, J = 14.0 Hz, 1H), 2.36 (d, J = 14.0 Hz, 1H), 1.66–1.32 (m, 10H), 1.27 (s, 3H), 0.97 (t, J = 7.1 Hz, 9H); 13C NMR (100 MHz, CD3OD) δ 175.2, 73.9, 72.8, 52.3, 48.3, 44.2, 41.6, 39.6, 37.2, 27.8, 22.1, 20.8, 15.3; HRESIMS (neg.) m/z 324.14903 [calcd for C13H26NO6S (M − H)−, Δ +0.40 mmu].
:30 to 100:0) and SiO2 column chromatography (Wakosil C-300, Wako Pure Chemical Industries, Ltd.; eluent, CH3Cl–MeOH, 95:5 to 0:100) to afford taurospongins B (1, 1.7 mg, 0.00048%, wet weight) and C (2, 4.2 mg, 0.0012%).
:1, 500 μl). After stirring for 12 h at 80 °C, the mixture was concentrated by Ar blowing. The residue was extracted with CHCl3, which was concentrated by Ar blowing to afford acetylenic fatty acid part (5, 0.5 mg, 1.28 μmol, 88%). The remaining CHCl3 insoluble material was taurineamide part (4, 0.4 mg, 1.23 μmol, 84%).:0 to 85:15) to afford 7 (1.81 g, 7.41 mmol, 90%); yellow oil; [α]21D − 28 (c 1.6, CHCl3); IR (neat) νmax 2957, 2877, 1738, 1458, 1223, 1005, 725 cm−1; 1H NMR (500 MHz, CDCl3) δ 4.58 (m, 1H), 4.34 (m, 1H), 2.67 (brd, J = 17.2 Hz, 1H), 2.43 (d, J = 17.2 Hz, 1H), 1.84 (m, 2H), 1.39 (s, 3H), 0.94 (t, J = 8.0 Hz, 9H), 0.60 (q, J = 8.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 170.2, 70.4, 66.1, 45.6, 36.9, 29.0, 6.8, 6.3; HRESIMS (pos.) m/z 267.13881 [calcd for C12H24O3NaSi (M + Na)+, Δ +0.12 mmu].:0 to 85:15) to afford 8 (1.81 g, 7.35 mmol, 97%), which was directly used in the next step; yellow oil; HRESIMS (pos.) m/z 269.15447 [calcd for C12H26O3NaSi (M + Na)+, Δ +0.13 mmu].:0 to 90:10) to afford 9 (1.29 g, 4.63 mmol, 62%), which was directly used in the next step; pale yellow oil; HRESIMS (pos.) m/z 301.13601 [calcd for C13H27O2ClNaSi (M + Na) +, Δ −0.10 mmu].:0 to 90:10) to afford 10 (1.56 g, 3.91 mmol, 84%), which was directly used in the next step; pale yellow oil; HRESIMS (pos.) m/z 421.19366 [calcd for C21H35O3ClNaSi (M + Na)+, Δ +0.04 mmu].:0 to 90:10) to afford mixture of 11a and 11b (1.21 g, 2.78 mmol, 79%), which was directly used in the next step; pale yellow oil; HRESIMS (pos.) m/z 457.27449 [calcd for C25H42O4NaSi (M + Na)+, Δ +0.03 mmu].:1, 30 mL) were added to the mixture, which was extracted with CH2Cl2. The organic layer was washed with brine, dried with MgSO4, and concentrated in vacuo. The residue was purified by a SiO2 column (n-hexane–EtOAc, 100:0 to 90:10) to afford 12 (1.16 g, 2.68 mmol, 95%); pale yellow oil; [α]22D + 5.0 (c 1.25, CHCl3); IR (neat) νmax 3019, 2876, 2211, 1673, 1455, 1377, 1240, 1171, 1114, 1045, 742, 698 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.37–7.26 (m, 5H), 4.75 (s, 2H), 4.60 (s, 2H), 3.72 (m, 2H), 2.61 (d, J = 17.1 Hz, 1H), 2.54 (d, J = 17.1 Hz, 1H), 2.50 (t, J = 7.3 Hz, 1H), 1.96 (ddd, J = 13.9, 7.3, 6.8 Hz, 1H), 1.89 (ddd, J = 13.9, 7.5, 6.4 Hz, 1H), 1.69 (qt, J = 7.3, 7.3 Hz, 2H), 1.37 (s, 3H), 0.95 (qt, J = 7.8 Hz, 9H), 0.93 (t, J = 7.3 Hz, 3H), 0.60 (q, J = 7.8 Hz, 6H); 13C NMR (150 MHz, CDCl3) δ 188.1, 138.0, 128.4, 127.8, 127.7, 94.7, 91.1, 82.6, 73.9, 69.4, 64.1, 47.4, 41.6, 33.8, 28.1, 17.6, 13.5, 7.0, 6.7; HRESIMS (pos.) m/z 455.25877 [calcd for C25H40O4NaSi (M + Na)+, Δ −0.04 mmu].:0 to 80:20) to afford 11a (563 mg, 1.30 mmol, 97%, 97:3 dr); pale brown oil; [α]23D + 4.8 (c 1.23, CHCl3); IR (neat) νmax 3441, 2956, 2875, 1455, 1376, 1150, 1111, 1041, 733 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.76 (s, 2H), 4.61 (s, 2H), 4.33 (brt, J = 6.5 Hz, 1H), 3.74 (t, J = 7.4 Hz, 2H), 2.44 (dd, J = 16.4 and 1.5 Hz, 1H), 2.36 (dd, J = 16.4 and 1.8 Hz, 1H), 2.03–1.75 (m, 3H), 1.63 (m, 2H), 1.46 (m, 2H), 1.33 (s, 3H), 0.94 (t, J = 7.9 Hz, 9H), 0.93 (t, J = 7.3 Hz, 3H), 0.59 (q, J = 7.9 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 137.8, 128.4, 127.9, 127.7, 94.5, 83.4, 82.3, 74.0, 69.3, 64.4, 62.4, 41.2, 40.1, 33.4, 28.0, 18.4, 13.7, 7.0, 6.7; HRESIMS (pos.) m/z 457.27512 [calcd for C25H42O4NaSi (M + Na)+, Δ +0.66 mmu].:0 to 2:1) to afford (S)-MTPA ester of 11a (0.7 mg, 1.08 mmol, 94%); 1H NMR (600 MHz, CDCl3) δ 7.71–7.29 (m, 10H), 5.56 (t, J = 6.7 Hz, 1H), 4.73 (s, 2H), 4.59 (s, 2H), 3.71 (t, J = 7.3 Hz, 2H), 3.58 (s, 3H), 2.46 (d, J = 16.4 Hz, 1H), 2.38 (d, J = 16.4 Hz, 1H), 1.93 (m, 1H), 1.86 (m, 1H), 1.73 (m, 2H), 1.35 (m, 2H), 1.31 (s, 3H), 0.93 (t, J = 7.9 Hz, 9H), 0.87 (t, J = 7.3 Hz, 3H), 0.57 (q, J = 7.9 Hz, 6H); HRESIMS (pos.) m/z 673.31519 [calcd for C35H49O6F3NaSi (M + Na)+, Δ +0.92 mmu].:3 dr) by using [(1R,2R)-N-(p-toluenesulfonyl)-1,2-duphenylethanediamine]-(p-cymene)ruthenium(II) through the same procedure as described for preparation of 11a; pale brown oil; [α]23D + 8.4 (c 1.77, CHCl3); IR (neat) νmax 3447, 2956, 2875, 1455, 1376, 1150, 1112, 1041, 741 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.76 (s, 2H), 4.61 (s, 2H), 4.33 (brs, 1H), 3.74 (t, J = 7.3 Hz, 2H), 2.44 (dd, J = 16.4 and 1.8 Hz, 1H), 2.36 (dd, J = 16.4 and 1.8 Hz, 1H), 1.98 (m, 1H), 1.87 (m, 2H), 1.63 (m, 2H), 1.46 (m, 2H), 1.33 (s, 3H), 0.94 (t, J = 7.8 Hz, 9H), 0.93 (t, J = 7.4 Hz, 3H), 0.59 (q, J = 7.8 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 137.8, 128.4, 127.9, 127.7, 94.5, 83.4, 82.3, 74.0, 69.2, 64.4, 62.4, 41.2, 40.1, 33.4, 28.0, 18.5, 13.7, 7.1 6.6; HRESIMS (pos.) m/z 457.27408 [calcd for C25H42O4NaSi (M + Na)+, Δ −0.38 mmu].:0 to 90:10) to afford 13a (675 mg, 1.23 mmol, 95%); colorless oil; [α]24D − 14.6 (c 1.17, CHCl3); IR (neat) νmax 2956, 2876, 1456, 1251, 1111, 1041, 836, 776, 731 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.38–7.26 (m, 5H), 4.75 (s, 2H), 4.60 (s, 2H), 4.33 (brt, J = 6.3, 1H), 3.73 (t, J = 7.4 Hz, 2H), 2.43 (dd, J = 16.4 and 1.5 Hz, 1H), 2.34 (dd, J = 16.4 and 1.9 Hz, 1H), 1.98 (m, 1H), 1.86 (m, 1H), 1.61 (m, 2H), 1.42 (m, 2H), 1.33 (s, 3H), 0.94 (t, J = 7.9 Hz, 9H), 0.91 (t, J = 7.3 Hz, 3H), 0.90 (s, 9H), 0.58 (q, J = 7.9 Hz, 6H), 0.11 (s, 3H), 0.09 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 137.9, 128.4, 127.9, 127.6, 94.6, 84.0, 81.1, 74.1, 69.2, 64.4, 62.9, 41.1, 41.1, 33.5, 28.0, 25.8, 18.6, 18.2, 13.8, 7.1, 6.7, −4.5, −5.1; HRESIMS (pos.) m/z 571.36206 [calcd for C31H56O4NaSi2 (M + Na)+, Δ +1.13 mmu].:0 to 90:10) to afford 15a (2.0 mg, 0.0092 mmol, 77%); colorless oil; [α]18D − 5.9 (c 2.02, CHCl3); IR (neat) νmax 3410, 2923, 2853, 1717, 1562, 1456, 1240, 1129, 774 cm−1; 1H NMR (400 MHz, CDCl3) δ 5.05 (brs, 1H), 3.64 (m, 1H), 2.56 (d, J = 15.6 Hz, 1H), 2.47 (d, J = 15.6 Hz, 1H), 1.61–1.29 (m, 10H), 1.28 (s, 3H), 0.92 (t, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 175.8, 71.5, 71.4, 44.6, 41.6, 39.6, 37.2, 26.7, 19.9, 18.8, 14.1; HRESIMS (pos.) m/z 241.14118 [calcd for C11H22O4Na (M + Na)+, Δ +0.15 mmu].:0 to 50:50) to afford 6a (19.0 mg, 0.058 mmol, 53%); colorless oil; [α]20D + 3.5 (c 1.14, MeOH); IR (neat) νmax 3366, 2955, 2871, 2280, 1638, 1555, 1284, 1144, 1059, 938 cm−1; 1H NMR (600 MHz, CD3OD) δ 3.67 (brt, J = 6.7 Hz, 2H), 3.58 (m, 1H), 3.03 (t, J = 6.6 Hz, 2H), 2.43 (d, J = 14.0 Hz, 1H), 2.36 (d, J = 14.0 Hz, 1H), 1.66–1.32 (m, 10H), 1.27 (s, 3H), 0.97 (t, J = 7.1 Hz, 9H); 13C NMR (100 MHz, CD3OD) δ 175.2, 73.9, 72.8, 52.3, 48.3, 44.2, 41.6, 39.6, 37.2, 27.8, 22.1, 20.8, 15.3; HRESIMS (neg.) m/z 324.14903 [calcd for C13H26NO6S (M − H)−, Δ +0.40 mmu].Antimicrobial assay
Antimicrobial assay of 1–3 against Escherichia coli, Staphylococcus aureus, Bacillus subtilis, Micrococcus luteus, Aspergillus niger, Candida albicans, Cryptococcus neoformans, and Trichophyton mentagrophytes was carried out as previously described.14 Amphotericin B, micafungin, hygromycin B, and kanamycin showed antifungal activity against Cryptococcus neoformans (MIC, <0.05, <0.1, 4.0, and 8.0 μg mL−1, respectively).Cytotoxic assays
Human epidermoid carcinoma (KB) and murine leukemia L1210 cells were cultured in an incubator at 37 °C for 48 h in 100 μl of medium containing various concentrations of test compounds dissolved in 1% DMSO. The IC50 values were obtained by plotting the logarithm of the concentration of the test compound versus the growth rate of the treated cells. Paclitaxel was used as positive control (IC50, <0.005 and <0.1 μg mL−1, respectively).Acknowledgements
We thank Mr Z. Nagahama and Mr K. Uehara, Okinawa, for their help with sponge collection, and Ms S. Oka, Instrumental Analysis Division, Equipment Management Center, Creative Reseach Institution, Hokkaido University, for measurements of mass spectrometry. We specially thank ADEKA corporation for supplying D-mevalonolactone, and Prof. Y. Iwabuchi, Graduate School of Pharmaceutical Sciences, Tohoku University, for supplying AZADO. This work was supported by The Naito Foundation, Cooperative Research Program of Medical Mycology Research Center, Chiba University, and Grant-in-Aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.Notes and references
- J. Kobayashi, T. Madono and H. Shigemori, Tetrahedron, 1995, 51, 10867 CrossRef CAS , and reference cited therein.
- D. Mori, Y. Kimura, S. Kitamura, Y. Sakagami, Y. Yoshioka, T. Shintani, T. Okamoto and M. Ojika, J. Org. Chem., 2007, 72, 7190 CrossRef CAS PubMed.
- A. A. Salim, J. Rae, F. Fontaine, M. M. Conte, Z. Khalil, S. Martin, R. G. Parton and R. J. Capon, Org. Biomol. Chem., 2010, 8, 3188 CAS.
- H. Ishiyama, M. Ishibashi, A. Ogawa, S. Yoshida and J. Kobayashi, J. Org. Chem., 1997, 62, 3831 CrossRef CAS.
- M. Shibuya, M. Tomizawa, I. Suzuki and Y. Iwabuchi, J. Am. Chem. Soc., 2006, 128, 8412 CrossRef CAS PubMed.
- K. Matsumura, S. Hashiguchi, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1997, 119, 8738 CrossRef CAS.
- I. Ohtani, T. Kusumi, Y. Kashman and H. Kakisawa, J. Am. Chem. Soc., 1991, 113, 4092 CrossRef CAS.
- W. M. Pearlman, Tetrahedron Lett., 1967, 8, 1663 CrossRef.
- J. R. Parikh and W. von E. Doering, J. Am. Chem. Soc., 1967, 89, 5505 CrossRef CAS.
- B. S. Bal, W. E. Childers Jr and H. W. Pinnick, Tetrahedron, 1981, 37, 2091 CrossRef CAS.
- M. Kunishima, C. Kawachi, F. Iwasaki, K. Terao and S. Tani, Tetrahedron Lett., 1999, 40, 5327 CrossRef CAS.
- Wu. Boshen, A. Mallinger and J. Robertson, Org. Lett., 2010, 12, 2818 CrossRef PubMed.
- J. H. Christopher, S. Yamanoi and S. V. Ley, Org. Biomol. Chem., 2003, 10, 1664 Search PubMed.
- H. Nagai, Y. Mikami, K. Yazawa, T. Gonoi and T. Yasumoto, J. Antibiot., 1993, 46, 520 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of natural and synthetic compounds. See DOI: 10.1039/c3ra47796g |
| This journal is © The Royal Society of Chemistry 2014 |