
Nitrogen functionalized graphite nanofibers/Ir nanoparticles for enhanced oxygen reduction reaction in polymer electrolyte fuel cells (PEFCs)
Abstract
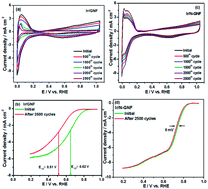
Nitrogen functionalization of graphite nanofibers (N-GNF) was performed using hexa methyl tetra amine (HMTA) as the nitrogen source and used as a support material for metal nanoparticle deposition. The successful incorporation of nitrogen was confirmed using X-ray photoelectron spectroscopy (XPS) and Raman spectroscopy analysis. Iridium (Ir) nanoparticles with a particle size of ∼2.2 nm were deposited onto N-GNF by a simple ethanol reduction method. The oxygen reduction reaction (ORR) activity of N-GNF and the ameliorating effect of ORR on Ir deposited N-GNF (Ir/N-GNF) were studied by various physicochemical and electrochemical methods. The enhancement of ORR activity for Ir/N-GNF was evidenced by high onset potentials and mass activities. The presence of nitrogen in the Ir/N-GNF catalyst facilitates quick desorption of the –OH species from the Ir surface and accelerates the electrochemical reaction of Ir particles which in turn enhances the ORR activity. The electrochemical stability of the Ir/N-GNF was investigated by repeated potential cycling up to 2500 cycles and was found to have excellent stability for ORR activity. The PEFC with Ir/N-GNF catalyst delivers a peak power density of 450 mW cm−2 at a load current density of 1577 mA cm−2, while the PEFC with Ir/GNF catalyst delivers a peak power density of only 259 mW cm−2 at a load current density of 1040 mA cm−2 under identical operation conditions.
 Please wait while we load your content...
Please wait while we load your content...

