
Unexpected different chemoselectivity in the aerobic oxidation of methylated planar catechin and bent epicatechin derivatives catalysed by the Trametes villosa laccase/1-hydroxybenzotriazole system†
Abstract
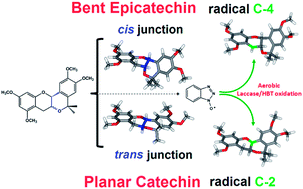
Unreported methylated catechin and epicatechin derivatives 5 and 6 were synthesized by an oxa-Pictet-Spengler reaction. Catechin 5 shows the B and C rings coplanar because of the formation of a trans junction between the C ring and the newly generated six-term cycle D, in turn condensed to ring B. In contrast, epicatechin 6 presents a bent geometry due to the establishment of a cis junction between the C ring and the newly formed cycle D. The oxidation of compounds 5 and 6 in the presence of the Trametes villosa laccase/1-hydroxybenzotriazole (HBT) system was investigated under aerobic conditions in both a biphasic system and a reverse micelle. The unexpected different chemoselective oxidation at the benzylic position of catechin and epicatechin derivatives 5 and 6 has been rationalized using a molecular modelling approach. These results demonstrate that the Trametes villosa laccase/HBT system represents a useful tool to functionalize the C-2 or C-4 position of phenolic compounds depending on the structural features.
 Please wait while we load your content...
Please wait while we load your content...

