Paper microfluidic extraction and direct smartphone-based identification of pathogenic nucleic acids from field and clinical samples
Christopher F. Fronczeka,
Tu San Parkb,
Dustin K. Harshmana,
Ariana M. Nicolinia and
Jeong-Yeol Yoon*ab
aBiomedical Engineering Graduate Interdisciplinary Program and Department of Biomedical Engineering, The University of Arizona, Tucson, AZ 85721, USA. E-mail: jyyoon@email.arizona.edu; Tel: +1-520-621-3587
bDepartment of Agricultural and Biosystems Engineering, The University of Arizona, Tucson, AZ 85721, USA
First published on 5th February 2014
Abstract
A rapid, paper microfluidic- and smartphone-based protocol was developed for the extraction and direct fluorescent identification of the nucleic acids of Salmonella Typhimurium from field and clinical samples. Initially, liquid samples (10% diluted) from fresh poultry packaging were loaded on the paper chips and were lysed with Tris–EDTA (TE) buffer. Nucleic acids from the lysed samples were eluted through the paper channel with TE buffer and the paper channel was excised into three pieces for the further polymerase chain reaction (PCR) assay. The extraction efficiency was determined by measuring fluorescence reflectance with either a benchtop optical detection system (consisting of an LED light source, a pair of optical fibers, and a miniature spectrophotometer, all built on micro-positioning stages) or a smartphone-based fluorescent microscope (in-house fabricated). The limit of detection of Salmonella Typhimurium in 10% poultry packaging liquid with cellulose paper was 103 CFU mL−1, while that extracted with nitrocellulose paper was 104 CFU mL−1 (as determined by both PCR and fluorescence reflectance). Cellulose channels proved more appropriate for measuring low and very high concentrations of pathogen DNA, while nitrocellulose proved better for analysing the mid-range concentrations. We observed that DNA migrated through nitrocellulose at a faster rate and further than through cellulose due to charge–charge repulsion between nitrocellulose and DNA (both negatively charged), thus contributing to consistent and efficient extraction. We tested the efficiency of Salmonella extraction from 10% poultry packaging liquid, 10% whole blood, and 10% fecal samples, and obtained comparable extraction efficiency, as confirmed by smartphone-based direct fluorescent detection. This protocol is suitable for the direct detection of total bacteria count in a dirty sample (when specificity is not necessary) as well as determining extraction efficiency. This protocol is compatible with PCR, to provide specific information about the type of pathogen present in sample.
Introduction
In this modern age, there has been a paradigm shift from reactive medicine to proactive medicine, and this general model includes the focus of on-site, rapid diagnostics. However, despite the advancements in field-deployable pathogen detection and point-of-care diagnostics, there is still an unacceptable level of outbreaks of common pathogens even in developed countries, originating from contaminated food, water, and air. These pathogens are often well-studied and can be very easily avoided if proper care is taken, and proper diagnostic tools would aid in the prevention of these illnesses. Many common bacterial infections, often originating from improper handling of food, lead to a large percent of hospitalizations and sometimes lead to death. These diseases cost the food and medical industry hundreds of millions of dollars per year. According to the U.S. Centers for Disease Control and Prevention (CDC), Salmonella species, among them Salmonella Typhimurium, affect nearly 50 million Americans each year and lead to the highest number of hospitalizations.1Clearly, more emphasis on on-site pathogen detection is necessary. In the past several years, much research has focused on developing on-site portable assays that maximize specificity and sensitivity while minimizing operation complexity. While the introduction of enzyme-linked immunosorbent assay (ELISA) has made great strides in sensitivity and specificity, it requires several washing steps and is not user-friendly.2 Today, the gold standard for the detection of a specific pathogen is to identify its specific nucleic acid sequence. However, since clinically relevant samples often contain very small amounts of DNA from pathogens, sequence amplification using polymerase chain reaction (PCR) is necessary.3 PCR is generally preferable over typical immunoassays due to better sensitivity and specificity. Unfortunately, PCR is quite labor intensive and requires sample preparation, nucleic acid extraction and purification, amplification, and product identification (typically through gel electrophoresis) – the whole process takes 4–6 hours (not counting the delivery time to a laboratory). In order to lower the total assay time, various miniature devices have been established such as PCR-on-a-chip4–8 and microdroplet manipulation.9,10 Also, cell culture has been introduced into PCR in order to increase the concentration of nucleic acid, towards decreasing the limit of detection or the number of cycles needed for PCR.11 However, these protocols focus primarily on amplification and do not account for laborious sample preparation, extraction, and detection. Generally, both ELISA and PCR require several hours of bench work, use expensive equipment, and are still laborious. Therefore, a more rapid, simple, and user-friendly apparatus that allows for sensitive and on-site pathogen detection should be developed. Because of its specificity, nucleic acid should be targeted, and a simple extraction device could provide fast sample preparation for PCR while providing a platform for preliminary detection. Direct DNA detection in field-deployable or point-of-care format has been attempted, particularly in lateral flow assay format,12,13 but these techniques require sample pre-treatment (including heat denaturation), may not be compatible with real samples, often target ribosomal RNA (rRNA) but not genomic DNA, and rely on colorimetric detection. Clearly, there is a need to directly target genomic DNA with a simplified protocol.
Paper microfluidics, an emerging field, has the potential to provide a cheap, user-friendly format for on-site detection of various pathogens from complex sample matrices. Paper microfluidic techniques have been utilized to detect glucose,14 proteins,15 and as an ELISA platform.16 In a recent study, a particle immunoassay for Salmonella detection was achieved on cellulose paper using a smartphone, and a 10 CFU mL−1 detection limit was achieved.17 Smartphones have recently been interfaced with a variety of biosensors for the optical detection of various hormones,18 parasites,19 and bacteria.20 In this study, we develop an easy-to-use paper microfluidic chip to extract and isolate genomic nucleic acid (DNA) of Salmonella Typhimurium from real field samples (which may be used in conjunction with portable or on-chip PCR) and propose a one-step direct detection apparatus (no PCR) using a smartphone.
Materials and methods
Salmonella Typhimurium
The Salmonella Typhimurium Z005 strain (ZeptoMetrix, Buffalo, NY, USA) was cultured in 25 mg mL−1 brain heart infusion broth (Remel, Lenexa, KS, USA). It was incubated for 12 hours at 37 °C with the final concentration of 8.6 × 108 CFU mL−1. This bacterial culture (assumed 109 CFU mL−1) was used to make serial dilutions in deionized (DI) water. The bacteria were re-cultured daily.Poultry packing liquid
Fresh chicken breasts and thighs were purchased from local stores and the fresh liquid was collected from the packaging with no further processing. The collected liquid samples were confirmed to be negative for Salmonella Typhimurium K005 using standard cell plating techniques and PCR (details discussed in “Conventional PCR for verification” section). The raw samples were then used to make 10% dilutions in DI water. Poultry packaging liquid samples were stored in refrigeration and used for up to 4 weeks. For testing, the sample matrices were spiked with the serially diluted Salmonella Typhimurium, while ensuring that the sample matrix was not significantly diluted further. The spiked samples were placed in 2 mL centrifuge tubes at room temperature during testing.Whole blood
Whole human blood, type O, with heparin sulfate, was purchased from the Interstate Blood Bank (Interstate Blood Bank Inc., Memphis, TN, USA). Blood aliquots were diluted to 10% in DI water. Samples were stored in refrigeration for up to 4 weeks. For testing, the samples were spiked with the serially diluted Salmonella at 105 CFU mL−1.Fecal sample
Fecal samples from an African spurred tortoise, swabbed from the floor of its habitat, were collected and placed in a centrifuge tube containing DI water. Further dilutions were necessary to achieve 10% dilution. Fecal samples were tested for the presence of Salmonella Typhimurium for negative confirmation. Samples were stored in refrigeration for up to 4 weeks. For testing, the samples were spiked with the serially diluted Salmonella at 105 CFU mL−1.Fabrication of paper microfluidic chips
Microfluidic channels were patterned on paper using a standard lithography technique with UV development.15 Channels were designed using SolidWorks 2010 (SolidWorks Corp., Concord, MA, USA) and were printed on clear transparency film to form a mask. Whatman cellulose #1 and nitrocellulose paper (Whatman Ltd., Kent, UK) strips were immersed in thinned SU-8 2075 negative photoresist and were UV-exposed with the mask following pre-baking. The paper chip was rinsed with acetone and isopropyl alcohol (Sigma-Aldrich, St. Louis, MO, USA), and the resulting hydrophilic channel consisted of a square loading site (7 mm × 7 mm), a channel for filtration (4 mm × 30 mm) and an absorbent pad (Fig. 1). All chips were kept in a sterile environment and free of dust particles. | ||
| Fig. 1 Top: patterned cellulose channel using modified lithography with equally-spaced elution regions. Bottom: the overall sample preparation is as follows: (a) load sample on paper; (b) add TE lysis buffer and incubate for 3 minutes; (c) add TE elution buffer and Qubit intercalating dye; (d) elute nucleic acids and measure fluorescence reflectance signal with a smartphone fluorescent microscope; optional: (e) excise specific paper regions, rinse the excised papers with TE buffer and load to a PCR reaction mixture. The flow shows theoretical separation of various components within paper. | ||
Sample loading onto paper microfluidic chips
Aliquots of cultured and serially diluted Salmonella Typhimurium, in DI water, 10% poultry packaging liquid, 10% whole blood, and 10% fecal samples, were loaded on the inlets of paper strips (Fig. 1a), and Tris–EDTA (TE) buffer (pH 8.0; Sigma-Aldrich) was loaded for lysis (Fig. 1b). The EDTA portion of TE buffer is known to lyse Gram negative bacteria, including Escherichia and Salmonella species, without enzymatic treatment (Gram positive bacteria typically require enzymatic treatment).21 Samples were allowed to incubate for 3 minutes, followed by an elution in TE buffer (Fig. 1c). At this point, the eluted sample was either loaded for conventional PCR, or the entire chip was analysed using fluorescent reflectance detection.Direct fluorescent detection with benchtop reader system
For direct fluorescent detection, the chip containing eluted nucleic acid was fastened to a reader system (Fig. 2), fabricated using a Dimension uPrint 3-D printer (Stratasys, Inc., Eden Prairie, MN, USA). 2 μL of Qubit fluorescent intercalating dye (Invitrogen Life Technologies, Carlsbad, CA, USA) was loaded at 3 pre-determined locations along the paper channel. The dye loading and chip imaging were performed in the dark using a benchtop positioning stage with a 475 nm LED light source (LS-450; Ocean Optics, Dunedin, FL, USA) and a miniature spectrophotometer (USB4000; Ocean Optics). Fiber optic cables (Ocean Optics) were used to guide the blue excitation light from the LED light source perpendicular to the chip regions of interest, and the green emission light to the miniature spectrophotometer (USB4000), collected at a pre-determined 85° angle from the chip, or 5° from the excitation. This angle was determined to minimize the direct reflectance from the paper while receiving maximum fluorescent emission from it. Salmonella in DI water, 10% poultry packaging liquid, 10% whole blood, or 10% fecal samples were loaded onto cellulose and nitrocellulose paper chips, and lysis and elution were carried out using TE buffer. Without allowing the paper chips to dry, Qubit dye (this is an intercalating dye that binds to double stranded DNA and subsequently fluoresce, just like SYBR Green dye, but much more sensitive) was loaded onto the three testing regions, and direct fluorescence detection was performed on the aforementioned benchtop reader system. To switch between detection positions, the holder was moved horizontally along a track so that the incident and detection angles remained constant. Signals were normalized to a negative control consisting of DI water and dye (or 10% sample matrix and dye), and standard curves were constructed from the data.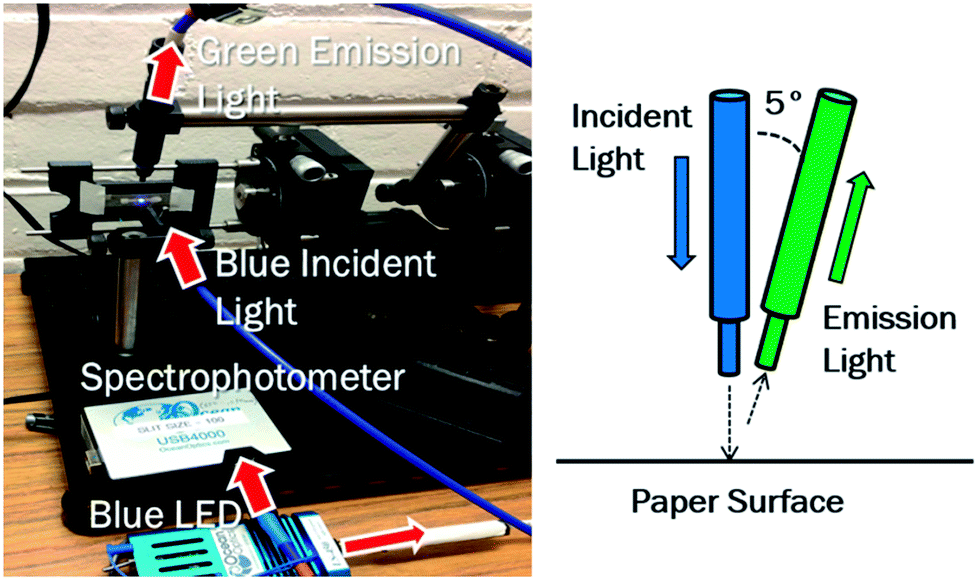 | ||
| Fig. 2 The benchtop reader system consists of a paper chip holder, a pair of optical fibers, a blue LED light source, a miniature spectrophotometer, all mounted on micro-positioning stages. The holder moves along a horizontal track to position the optical fibers at three different positions of the paper channel. Various detection angles were tested, and 5° from the incident light was found to minimize the direct reflectance from the paper while receiving maximum fluorescent emission from it. | ||
Direct fluorescent detection with smartphone
Fluorescent signals were detected on the patterned paper chips using an iPhone 4 (Apple Inc., Cupertino, CA, USA) and a fluorescent miniature microscope attachment, designed and fabricated in our laboratory (Fig. 3). The scope contains two 10× objective lenses for focus (catalog number LA1560-A; Thor Labs, Newton, NJ, USA), 492 nm bandpass filter (catalog number 65-087; Edmund Optics Inc., Barrington, NJ, USA), 520 nm bandpass filter (catalog number 65-093; Edmund Optics), and 500 nm dichroic shortpass filter (catalog number 69-178; Edmund Optics). A blue LED was used to create lighting, and the CMOS camera of a smartphone captured the high resolution images (5 megapixel). Briefly, three concentrations of Salmonella were spiked into 10% poultry packaging liquid, and lysis and elution were performed using TE buffer in cellulose and nitrocellulose paper chips. Qubit intercalating dye was added to the optimal position 2 on the chip. Images were taken with the iPhone 4 and the aforementioned smartphone-based fluorescent miniature microscope. ImageJ software (U.S. National Institutes of Health, Bethesda, MD, USA) was used to determine the green pixel intensity, and these values were used to form a standard curve normalized to the negative control (the 10% poultry packaging liquid without Salmonella). Experiments were replicated in triplicate. In addition, 10% whole blood and 10% fecal samples were tested in addition to the 10% poultry packaging liquid. These additional samples were spiked with 105 CFU mL−1 Salmonella, and data points were normalized to a negative control for each sample. Again, testing in both cellulose and nitrocellulose was in triplicate. | ||
| Fig. 3 A smartphone with a fluorescent miniature microscope attachment directly measures the fluorescent reflectance from a composite chip, consisting of four separate paper chips. Measurements were made separately for each channel. The fluorescent miniature microscope includes two bandpass filters, two 10× objective lenses, a dichroic mirror, and a blue LED (475 nm). | ||
Conventional PCR for verification
Target nucleic acid was eluted from the paper channels by excising specific portions of the channel, followed by dipping in and rinsing with TE buffer. The resulting bacterial genomic DNA was amplified with conventional PCR, using 2× Promega Green Master Mix (Promega Corporation, Seattle, WA, USA), for 35 cycles on a conventional thermocycler (MJ Research, Waltham, MA, USA). The following primer sets were used to target the Salmonella Typhimurium Z005 genome: sal201F (5′-CGGGCCTCTTGCCATCAGGTG-3′) and sal597R (5′-CACATCCGACTTGACAGACCG-3′). Standard gel electrophoresis with ethidium bromide fluorescent staining was used for amplicon determination. Cured 2% agarose gel (Sigma-Aldrich) was immersed in 1× Tris–acetate–EDTA (TAE) buffer (Invitrogen Life Technologies), and a power supply provided 120 V and 0.1 A for 45 minutes for electrophoresis (Thermo Fischer Scientific; Waltham, MA, USA). Amplified samples were loaded into the gel along with 1 kbp DNA ladder (Invitrogen Life Technologies). The gel was soaked in ethidium bromide solution (Sigma-Aldrich) for 20 minutes and imaged using a Gel Doc 1000 imaging system (Bio-Rad Laboratories, Hercules, CA, USA).Results and discussion
Salmonella extraction from poultry packing liquid
Conventional PCR was used to verify that DNA was successfully extracted from Salmonella on cellulose and nitrocellulose paper channels (Fig. 4). Negative controls were 10% poultry packaging liquid, free of bacteria, loaded onto the chip. An elution step was performed at the three regions of interest on the paper, and the gel results were obtained for Salmonella at various concentrations in 10% poultry packaging liquid. Fig. 4 shows the positive gel bands (at the expected 400 bp product length) at the lowest detectable Salmonella concentrations with cellulose (a) and nitrocellulose paper chips (b). The results suggest a 103 CFU mL−1 detection limit in cellulose paper and a 104 CFU mL−1 detection limit in nitrocellulose paper, well below the infectious dose.22 | ||
| Fig. 4 Select results of PCR verification gels. Cellulose (a) and nitrocellulose (b) paper extraction, of Salmonella (103 CFU mL−1) and (104 CFU mL−1), respectively, spiked in 10% poultry packaging liquid, eluted at three regions along the paper channel. Bands occurred at the expected 400 bp product length. No bands were observed for <103 CFU mL−1 on cellulose and <104 CFU mL−1 on nitrocellulose extraction, indicating the detection limits. Negative control is with unspiked 10% poultry packaging liquid loaded. This set of experiments illustrates the movement of large target DNA (4.86 Mbp) was faster through nitrocellulose compared to cellulose. | ||
This difference in detection limit is due to the superior filtration properties of cellulose paper. In addition, motility of DNA through both paper types was observed. Gel results suggest that DNA migrates through nitrocellulose farther and faster than through cellulose channels. This is likely due to the fact that the Salmonella Typhimurium genome is quite large (4.87 Mbp), and DNA may be physically entrapped within cellulose fibers (a downside of superior filtration property of cellulose). However, DNA will not preferentially interact with nitrocellulose but will instead be repelled through charge–charge repulsion (both nitrocellulose and DNA are negatively charged).23 For this reason, target DNA will travel through nitrocellulose further than through cellulose. The data suggest that a majority of DNA is captured at region 2 in cellulose and region 3 (furthest section in channel from the loading site) in nitrocellulose. Both types of paper are suitable for extraction and direct detection due to their wettability.
Direct fluorescence detection with benchtop reader system
Towards creating a field-deployable diagnostic tool to (1) collect preliminary data on total bacteria count in a sample and/or (2) check for successful DNA extraction from a sample, fluorescence reflectance measurements were made using Qubit intercalating dye and a benchtop optical detection system (Fig. 2). In order to minimize interfering backscatter signal caused by the wet paper, the detection angle was set to 85° from the paper (5° from the incident light) in order to minimize backscatter from the paper (Fig. 2). Green fluorescence reflectance was measured using Qubit intercalating dye to determine DNA extraction efficiency from clean Salmonella culture (in DI water) using cellulose (Fig. 5a) and nitrocellulose paper (Fig. 5b), over three different regions of a paper channel. Similarly, DNA extraction efficiency was determined from Salmonella spiked in 10% poultry packaging liquid in cellulose (Fig. 6a) and nitrocellulose paper (Fig. 6b). Total assay time was 5 minutes.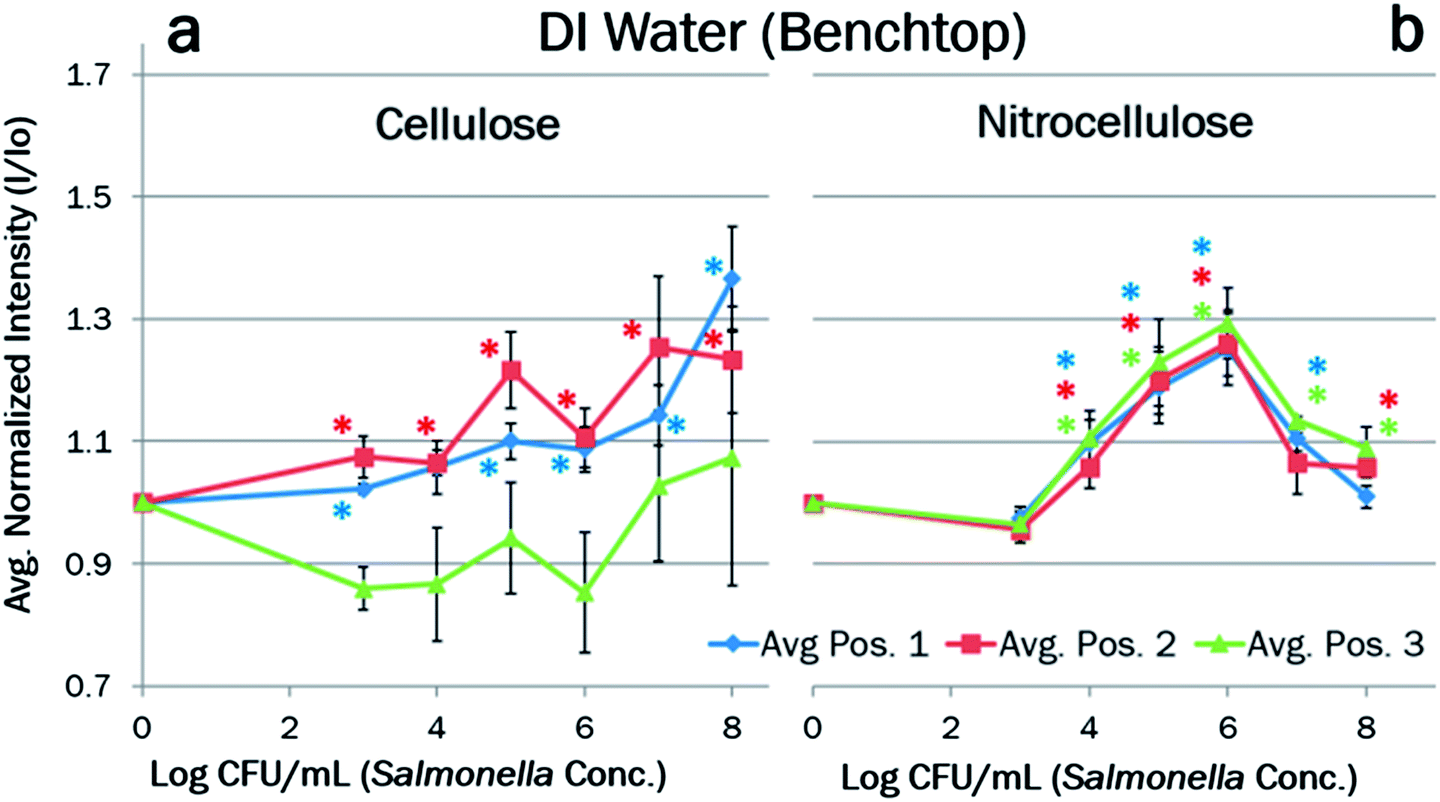 | ||
| Fig. 5 Fluorescent reflectance intensities taken on a benchtop reader system using Qubit intercalating dye, for the DNA extraction from clean Salmonella (in DI water) on (a) cellulose and (b) nitrocellulose paper chips. Samples were normalized to a negative control (DI water + Qubit intercalating dye) to create a standard curve (n = 3). Error bars represent standard errors. * indicates the data points with [average − 2 × standard error] being higher than 1, which are statistically significant over the negative controls with 95% confidence. | ||
 | ||
| Fig. 6 Fluorescent reflectance intensities taken on a benchtop reader system using Qubit intercalating dye, for the DNA extraction from Salmonella spiked in 10% poultry packaging liquid on (a) cellulose and (b) nitrocellulose paper chips. Samples were normalized to a negative control (10% poultry packaging liquid with no bacteria present + Qubit intercalating dye) to create a standard curve (n = 3). Error bars represent standard errors. * indicates the data points significantly different over the negative controls with 95% confidence. | ||
Standard curves, normalized to DI water (Fig. 5) and the negative control of 10% poultry packaging liquid (Fig. 6), showed a general increase in normalized intensity with increasing pathogen concentration. Negative controls were taken for each set of experiments due to the variation in paper chips as well as experimental conditions. Cellulose extraction of clean Salmonella yield a detection limit of 103 CFU mL−1 (Fig. 5a), which is in good agreement with the PCR gel results (Fig. 4). These curves also show the average maximum signal intensity output over the range at position 2, which is also in concordance with the PCR gel data (Fig. 4). However, there is a dip in signal for positions 1 and 2 of cellulose paper chip around 106 CFU mL−1, which can be explained by Salmonella colonizing and therefore clogging the paper fibers. This colonization was consistently observed under a light microscope with 40× objective lens. The normalized fluorescent intensity starts to re-increase beyond 107 CFU mL−1, presumably because there are enough available cells present to yield DNA to bypass the clogging. This clogging may become a larger obstacle when using poultry packaging liquid due to the interfering components of the sample. However, the results shown in Fig. 6a (with 10% poultry packaging liquid) show even less apparent dips. Possible explanations include: (1) the standard curve was normalized to negative control comprised of the poultry packaging liquid free of bacteria, and (2) proteins from poultry packing liquid, especially albumin and phospholipids (debris of lysed cells), acted as stabilizing agents or natural surfactants that reduced additional clogging, thereby achieving sufficient extraction efficiency.
Nitrocellulose extraction of clean Salmonella and Salmonella spiked in 10% poultry packaging liquid yielded a detection limit of 104 CFU mL−1 (Fig. 5b and 6b), which is also in good agreement with the PCR gel results (Fig. 4). Unlike the results with cellulose chips, the curves are quite linear up to 106 CFU mL−1 with smaller error bars, followed by a decrease in very high Salmonella concentrations. Optimal detection occurs at position 3, although there is less variation in the data between the three positions than in cellulose. This different trend can be explained by the fact that the contents of samples do not penetrate and bind strongly into the nitrocellulose fibers nearly as well as they do into the cellulose fibers, and the DNA likely migrates over the paper to the opposite end of the strip due to charge–charge repulsion. While there is still sufficient mixing of sample and lysis buffer, extraction efficiency is reduced due to lack of paper filtration, thus yielding a higher detection limit than in cellulose. The peak intensities were ∼1.25 in DI water (Fig. 5b) and ∼1.42 in 10% poultry packing liquid (Fig. 6b), both at 106 CFU mL−1, which are much higher than those with cellulose chips at the same Salmonella concentration. Again, the presence of albumin and phospholipids in the poultry packaging liquid can be attributed to this reproducible and linear fluorescence reading, helping to break up colonies and thereby eliminating the clogging as a natural surfactant.
All data points reported are in triplicate (n = 3), and the error bars represent standard errors. Since all data points were normalized to the negative controls, we indicated the data points with [average − 2 × standard error = 95% confidence interval] being higher than 1 using * marks. For cellulose chips (Fig. 5a and 6a), many data points were significantly higher than then negative controls for positions 1 and 2 but not for position 3, while most points for all three positions were significantly higher than the negative controls for nitrocellulose chips (Fig. 5b and 6b). In general, the curves were more linear with smaller error bars with nitrocellulose than with cellulose chips, which can be explained by the variances in cellulose fiber threads, which were not found in nitrocellulose fibers. In higher concentrations on both cellulose and nitrocellulose paper, particularly 108 CFU mL−1 spiked in 10% poultry packaging liquid, large error bars and poor statistics stem from very high amounts of bacteria, protein, and colonization.
Because of the dip between 105 to 107 CFU mL−1 due to clogging, cellulose channels proved more appropriate for measuring low and very high concentrations of pathogen DNA. Above and below this concentration range, there is a good increase in fluorescence, but this dip may be problematic in obtaining a consistent reading. By contrast, there is no dip between 105 to 107 CFU mL−1 in nitrocellulose paper. The most consistent increase in fluorescence appears between 104 to 106 CFU mL−1, making nitrocellulose better for analysing the mid-range concentrations.
Direct fluorescence detection with benchtop reader system
Direct detection experiments were repeated on cellulose and nitrocellulose paper chips using Salmonella at three different concentrations spiked in 10% poultry packaging liquid, and fluorescent intensities were captured using a smartphone (iPhone 4) attached with a fluorescent miniature microscope developed in our laboratory. The fluorescent signal at the optimized position 2 was captured through the miniature microscope with the CMOS image detector of a smartphone, and pixel intensity was analysed on ImageJ software. A standard curve was constructed (averages from three different experiments), normalized to the same negative control, and the data closely corresponds to the data collected using the benchtop reader system (Fig. 7). 95% confidence intervals, [average − 2 × standard error], indicate the detection limit of 103 to 104 CFU mL−1. Similar to the other data observed on cellulose, a dip appeared at the expected 106 CFU mL−1, which is due to clogging prior to region 1 on the chip. Meanwhile, better linearity and smaller error bars were again observed with nitrocellulose chips, in accordance with the results with the benchtop reader system (Fig. 5 and 6). The total assay time remained 5 minutes. | ||
| Fig. 7 Fluorescent reflectance intensities taken on a smartphone-based fluorescent miniature microscope using Qubit intercalating dye, for the DNA extraction from Salmonella spiked in 10% poultry packaging liquid on (a) cellulose and (b) nitrocellulose paper chips. Samples were normalized to a negative control (refer to Fig. 6) to create a standard curve (n = 3). Error bars represent standard errors. * indicates the data points significantly different over the negative controls with 95% confidence. | ||
Salmonella (105 CFU mL−1) in 10% whole blood and 10% fecal sample were also extracted in both paper types and detected using the smartphone-based fluorescent miniature microscope. Signals were normalized to their own negative controls. All data points showed a good statistical significance (95% confidence) over the negative controls, indicating successful DNA extraction (Fig. 8). All three data shown in Fig. 8 show no significant difference, indicating the extraction efficiencies are similar over three different sample matrices.
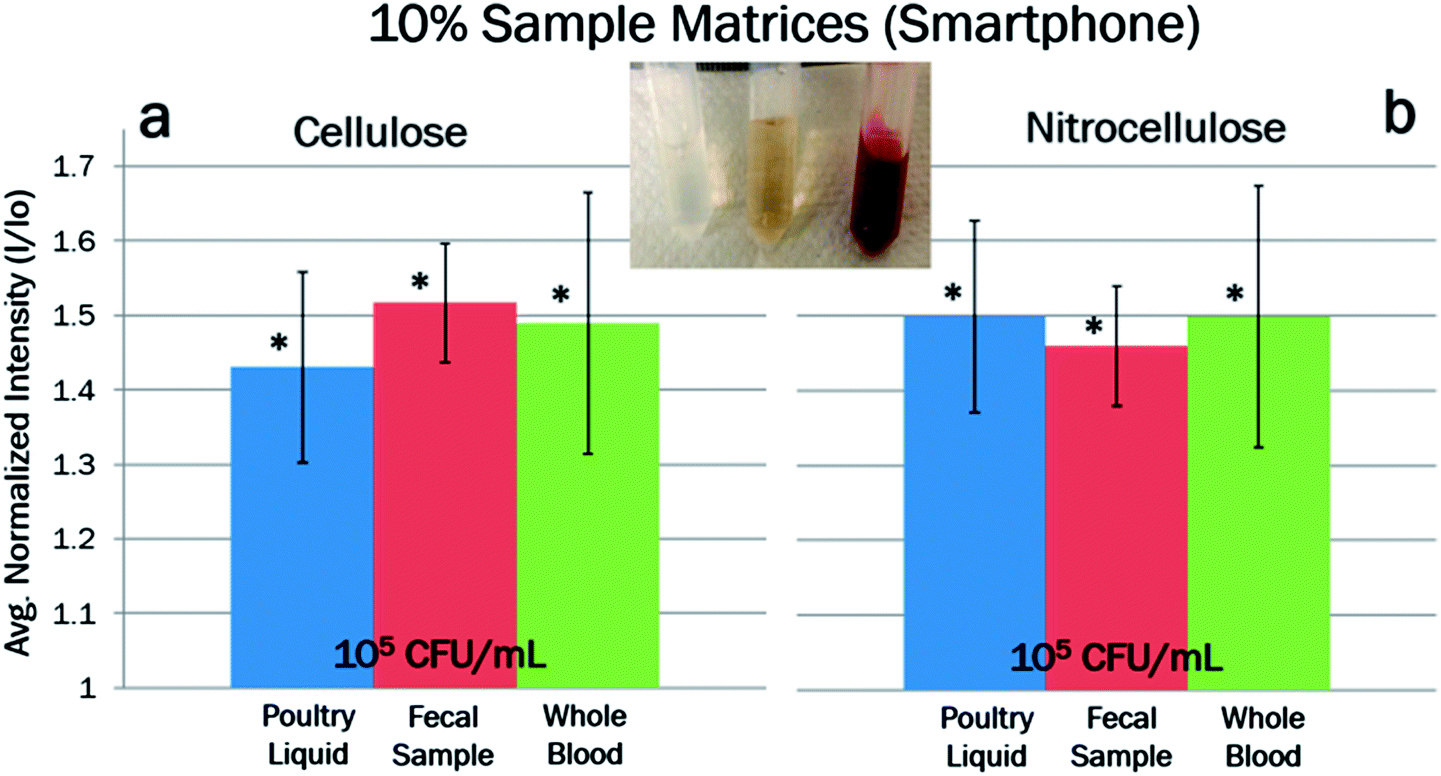 | ||
| Fig. 8 Fluorescent reflectance intensities taken on a smartphone-based fluorescent miniature microscope using Qubit intercalating dye, for the DNA extraction from Salmonella spiked in 10% poultry packaging liquid, 10% fecal sample, and 10% whole blood, on (a) cellulose and (b) nitrocellulose paper chips. Salmonella concentration was fixed at 105 CFU mL−1. Samples were normalized to a negative control (no bacteria present but with the same sample matrix) to create a standard curve (n = 3). Error bars represent standard errors. * indicates the data points significantly different over the negative controls with 95% confidence. | ||
Conclusions
We have created a sensitive, portable, low-cost device that provides a user-friendly method of extracting total bacterial genomic nucleic acid on a paper microfluidic format. Using fluorescence from Qubit intercalating dye, Salmonella Typhimurium Z005 in poultry packaging liquid was quantified in cellulose and nitrocellulose paper chips in two formats: benchtop optical detection system and smartphone-based fluorescent miniature microscope. The assay was then shown to work well with conventional PCR. This platform, along with the smartphone apparatus, will detail an accurate total bacterial count in a real sample while providing the flexibility of interfacing with PCR. This assay is more rapid than the gold standard and can be accomplished in-field in 5 minutes, and it is deposable due to the low cost of fabrication. The assay is sensitive and has a limit of detection well below the infectious dose of Salmonella. Cellulose channels proved more appropriate for measuring low and high concentrations of pathogen DNA, while nitrocellulose proved better for analysing the mid-range concentrations with improved linearity and reproducibility. The presence of three different relevant sample matrices did not impact the accuracy or reproducibility of the normalized intensity signal, nor does it affect PCR. This test has the potential to impact diagnostic worldwide due to its low cost, portability, and ability to interface with a smartphone. In the future, our system can be multiplexed by providing multiple channels on the paper chip for multiple tests, and an element of specificity can be implemented for sorting either DNA varieties or pathogen varieties to create a true field-deployable total analysis system. This system has the potential to have a major impact on the food and medical industries by enhancing field-deployable testing and point-of-care diagnostics and can save these industries millions of dollars by preventing the needless outbreak of common diseases.Acknowledgements
We thank everyone in Biosensors Laboratory at the University of Arizona, especially Scott Brechbiel for experimental assistance. This research was supported by Animal and Plant Quarantine Agency, South Korea (I-1541780-2012-13-0101).Notes and references
- U.S. Centers for Disease Control and Prevention (CDC), Trends in Foodborne Illness in the United States, 2012, http://www.cdc.gov/Features/dsFoodNet2012/index.html.
- J. Mairhofer, K. Roppert and P. Ertl, Sensors, 2009, 9, 4804 CrossRef CAS PubMed.
- F. C. Huang, C. S. Liao and G. B. Lee, Electrophoresis, 2006, 27, 3297 CrossRef CAS PubMed.
- M. U. Kopp, A. J. de Mello and A. Manz, Science, 1998, 280, 1046 CrossRef CAS.
- K. Y. Lien, W. C. Lee, H. Y. Lei and G. B. Lee, Biosens. Bioelectron., 2007, 22, 1739 CrossRef CAS PubMed.
- S. Takenaka, K. Yamashita, M. Takagi, Y. Uto and H. Kondo, Anal. Chem., 2000, 72, 1334 CrossRef CAS.
- T. de Lumley-Woodyear, C. N. Campbell and A. Heller, J. Am. Chem. Soc., 1996, 118, 5504 CrossRef CAS.
- J.-G. Lee, K. H. Cheong, N. Huh, S. Kim, J.-W. Choi and C. Ko, Lab Chip, 2000, 6, 886 RSC.
- D. J. You, P. L. Tran, H.-J. Kwon, D. Patel and J.-Y. Yoon, Faraday Discuss., 2011, 149, 159 RSC.
- D. K. Harshman, R. Reyes, T. S. Park, D. J. You, J.-Y. Song and J.-Y. Yoon, Biosens. Bioelectron., 2014, 53, 167 CrossRef CAS PubMed.
- D. Jonas, M. Speck, F. D. Daschner and H. Grundmann, J. Clin. Microbiol., 2002, 40, 1821 CrossRef CAS.
- X. Mao, Y. Ma, A. Zhang, L. Zhang, L. Zeng and G. Liu, Anal. Chem., 2009, 81, 1660 CrossRef CAS PubMed.
- S.-J. Lo, S.-C. Yang, D.-J. Yao, J.-H. Chen, W.-C. Tuc and C.-M. Cheng, Lab Chip, 2013, 13, 2686 RSC.
- E. Carrilho, A. W. Martinez and G. M. Whitesides, Anal. Chem., 2009, 81, 7091 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, E. Carrilho, S. W. Thomas III, H. Sindi and G. M. Whitesides, Anal. Chem., 2008, 80, 3699 CrossRef CAS PubMed.
- C.-M. Cheng, A. W. Martinez, J. Gong, C. R. Mace, S. T. Phillips, E. Carrilho, K. A. Mirica and G. M. Whitesides, Angew. Chem., Int. Ed., 2010, 122, 4881 CrossRef.
- T. S. Park, W. Li, K. E. McCracken and J.-Y. Yoon, Lab Chip, 2013, 13, 4832 RSC.
- D. J. You, T. S. Park and J.-Y. Yoon, Biosens. Bioelectron., 2013, 40, 180 CrossRef CAS PubMed.
- O. Mudanyali, S. Dimitrov, U. Sikora, S. Padmanabhan, I. Navruz and A. Ozcan, Lab Chip, 2012, 12, 2678 RSC.
- H. Zhu, I. Sencan, J. Wong, S. Dimitrov, D. Tseng, K. Nagashima and A. Ozcan, Lab Chip, 2013, 13, 1282 RSC.
- D. G. Pitcher, N. A. Saunders and R. J. Owen, Lett. Appl. Microbiol., 1989, 8, 151 CrossRef CAS.
- T. D. Lawley, D. M. Bouley, Y. E. Hoy, C. Gerke, D. A. Relman and D. M. Monack, Infect. Immun., 2008, 76, 403 CrossRef CAS PubMed.
- V. Munster, A. Wallensten, B. Olsen, G. F. Rimmelzwaan, A. D. M. E. Osterhaus and R. A. M. Fouchier, in Avian Influenza: Prevention and Control, ed. R. S. Schrijver and G. Koch, Springer, Dordrecht, 2005, ch. 4, pp. 25–30 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |