Self-assembly and phase separation of amphiphilic dyads based on 4,7-bis(2-thienyl)benzothiodiazole and perylene diimide†
Jiang Peng,
Feng Zhai,
Xinyan Guo,
Xinpeng Jiang and
Yuguo Ma*
Beijing National Laboratory for Molecular Sciences (BNLMS), Key Lab of Polymer Chemistry & Physics of Ministry of Education, College of Chemistry, Peking University, Beijing, 100871, China. E-mail: ygma@pku.edu.cn; Fax: +86 10-6275-1708; Tel: +86 10-6275-6660
First published on 20th January 2014
Abstract
To get highly ordered organic structures at the nanoscale, a series of new electronic donor–acceptor (D–A) dyads were synthesized. The dyads bearing different side chains (lipophilic or amphiphilic) or linkers (Long or Short) showed variable self-assembly behaviour. Long-linker dyads can fold in dilute solution, but the folding of short-linker dyads was not observed. Intermolecular D–A interactions and acceptor–acceptor (A–A) aggregation were proved to co-occur in chloroform for all four dyads. In the bulk state, amphiphilic dyads could overcome the intrinsic D–A interactions and achieve better D–A phase separation than lipophilic ones due to the incompatibility of the side chains. The dyad with a short-linker and amphiphilic side chains, Samphi, had the ability to form a gel, and both of the amphiphilic dyads could form nanofibres.
Introduction
In bulk heterojunction organic photovoltaic devices, bicontinuous arrays of electron donors and acceptors are a prerequisite.1 As an inherent property of organic semiconductors, the exciton diffusion length is ca. 5–10 nm.2 In traditional bulk heterojunction devices, donor and acceptor materials are blended with insufficient microphase separation, which results in exciton loss.3 To maximize the heterojunction interface, researchers paid much attention to small molecule dyads consisting of electron donor (i.e. p-type semiconductors) and electron acceptor (i.e. n-type semiconductors) units,4,5 which are promising for optimizing charge separation and charge transportation processes.To link electron donors and acceptors together, various linkages including soft alkyl chains,5a rigid phenylene acetylene,5b coordination bonding and hydrogen-bonding6 were used. Linkers are of key importance in dyads, since they affect the donor–acceptor (D–A) distance and D–A interface. However, the donors tend to stack with acceptors, forming charge transfer complexes that trap excitons.7 To induce D–A separation at the nanoscale and drive the formation of well-defined nano-morphology, incompatible wedges (e.g. dodecyl and triethylene glycol (TEG) chains5a,b) were equipped at each end of the amphiphilic dyads.4,5 The cooperativity of π–π stacking and incompatibility of the side chains enables the phase separation of both electron donors and acceptors.
In the present contribution, a series of new D–A dyads containing 4,7-bis(2-thienyl)-2,1,3-benzothiodiazole (T2BTZ) and perylene diimide (PDI) were synthesized (Fig. 1). A benzothiodiazole unit was incorporated to lower the LUMO level and red-shift the absorption onset,8 compared with the short-wavelength absorption (ca. 500 nm onset position) of commonly used oligothiophene.5a,b Incompatible triethylene glycol and dodecyl chains were utilized to improve phase separation (Samphi and Lamphi). The corresponding dyads with dodecyl chains on both sides (Slipo and Llipo) were synthesized as reference compounds. The independent donor (T2BTZ) and acceptor (PDI) were also prepared as reference compounds. Moreover, two amino acids were used to tune the distance between the donor and acceptor. The aliphatic linkers would avoid conjugated electron coupling between both parts.
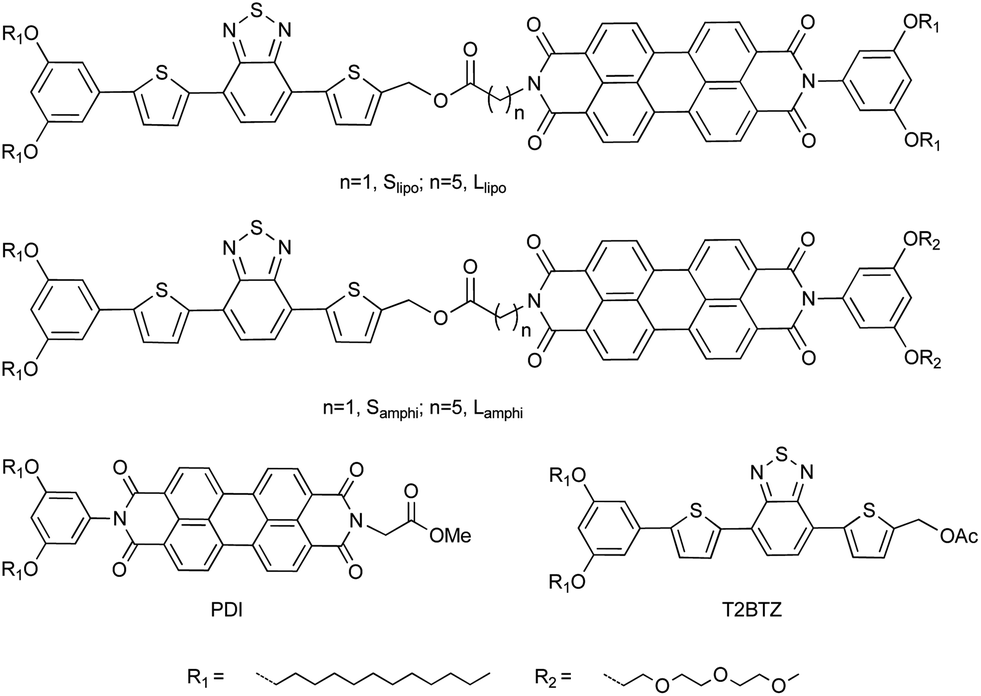 | ||
| Fig. 1 Chemical structures of four dyads and reference compounds. The donor and acceptor are 4,7-bis(2-thienyl)benzothiodiazole (T2BTZ) and perylene diimide (PDI), respectively. | ||
The influence of side chains and linker length on assembly behaviour, phase separation and nano-morphology was systematically investigated. Intramolecular D–A interaction of long-linker dyads was first observed in dilute solution. Intermolecular D–A interactions and A–A aggregation were both observed in concentrated solution. The dyads could achieve D–A phase separation assisted by the amphiphilic side chains, and the assembly morphologies differed with side chains and linker length. To the best of our knowledge, the linker length influence on dyad assembly and intramolecular D–A interactions has rarely been studied.9
Results and discussion
Self-assembly in solution
UV-visible spectroscopy was carried out to study the ground state electronic properties of the dyads in solution. Slipo and Samphi showed identical absorption, suggesting that the electronic properties of the donor and the acceptor are not influenced by the side chains. The spectra of Slipo and Samphi totally overlapped with the spectra summation of the reference compounds PDI and T2BTZ (Fig. 2a), demonstrating no electronic interaction between donor and acceptor (Fig. 2c). However, after the 0–0 transition peaks belonging to the acceptor component were normalized, Llipo and Lamphi had relatively higher 0–1 transition intensities than Slipo and Samphi, along with red-shifted onset wavelengths10 (Fig. 2b, black arrows). Based on this result, we proposed that in Llipo and Lamphi, T2BTZ may fold back to interact with PDI intramolecularly due to the flexibility of the long linker (Fig. 2c). To rule out the possibility of intermolecular aggregation, concentration-dependent UV-vis spectra of Llipo and Lamphi were recorded (Fig. S1†). The result confirmed that the spectral change indeed originated from intramolecular folding instead of intermolecular aggregation. Furthermore, the 0–1 transition intensity of Llipo is slightly higher than that of Lamphi (Fig. 2b, red arrow), and this happens at several selected concentrations from 10−6 M–10−5 M (Fig. S1†). We tentatively explain it as follows: when folding happens, the side chains on both sides approach each other intramolecularly. For Llipo, the affinity of the dodecyl chains induced folding of the D–A unit, while for Lamphi, the incompatibility of the dodecyl and triethylene glycol chains slightly inhibited folding. As far as we know, this is the first report on PDI–T2BTZ interactions and intramolecular side chain incompatibility, characterized by absorption spectroscopy in dilute solution. | ||
| Fig. 2 (a) UV-visible absorption spectra of short-linker dyads and reference compounds; (b) normalized absorption spectra of short-linker and long-linker dyads (chloroform, 5 × 10−6 M); (c) schematic diagram of intramolecular folding of short-linker and long-linker dyads. | ||
To understand the assembly behaviour of the donor and acceptor segments in solution, all four dyads were studied by concentration-dependent 1H NMR in CDCl3 (Fig. 3). Taking Samphi as an example and diluting its CDCl3 solution, we found that the peaks of the PDI and T2BTZ protons moved downfield and the fine structure became clear (Fig. 3, black arrows), indicating the assembly of Samphi. Moreover, detailed evidence for assembly was found from the upfield chemical shifts (Fig. 3, red arrow) of protons a and b attached to PDI. Through chemical structural analysis, we propose that proton a forms a weak hydrogen bond with the nearby carbonyl oxygen in the aggregates, which leads to a lower chemical shift of proton a. As the solution was diluted, the aggregates slowly dissociated, enabling free rotation of the phenyl ring. Therefore, the hydrogen bond was weakened and the chemical shift of proton a moved upfield. Meanwhile, proton b had a slight upfield shift due to the electronic effect from a. These phenomena demonstrate that self-assembly of Samphi exists in chloroform at this concentration range (ca. 1 mM to 10 mM). The other dyads showed similar chemical shift changes when diluting their CDCl3 solutions (Fig. S7–S10†), indicating the occurrence of self-assembly.
 | ||
| Fig. 3 Molecular structure of Samphi and the concentration-dependent 1H NMR spectra of Samphi in CDCl3. | ||
To further investigate the assembly details, a 1H NMR dilution experiment of the reference compounds PDI and T2BTZ and a mixture of both compounds was carried out in CDCl3. The chemical shifts of the most downfield PDI proton (c) and T2BTZ proton (d) at four different concentrations (30 mM to 9 mM) are summarized in Fig. 4 (see the spectra in Fig. S7–S13†). The δ of proton c changed by a maximum value (Δδ) of 0.09 indicating that PDI can self-assemble. Proton d only had a Δδ value of 0.013, which indicates that the self-assembly tendency of T2BTZ is weak. However, the Δδ of proton d increased significantly (Δδ = 0.09) when PDI and T2BTZ were mixed together. It means that T2BTZ has a tendency to co-assemble with PDI. In the four dyads, the Δδ of proton d was apparently larger than 0.013 (Table S1†), so the D–A co-assembly existed. Therefore, it is necessary to introduce different side chains to overcome the D–A interactions in order to achieve phase separation.
 | ||
| Fig. 4 Selected proton chemical shift changes of PDI (blue triangle), T2BTZ (black square) and their equimolar mixture (purple inverted triangle for PDI and red circle for T2BTZ) studied by 1H NMR experiments. (CDCl3, 30 mM to 9 mM, 25 °C). | ||
Morphology control and phase separation
The active layer morphology is of paramount importance for organic semiconductor devices (OSCs, OFET, etc.).11 Herein, the morphology and phase separation, which were influenced by side chains, linkers and aromatic π–π interactions, were investigated.Since all four dyads had assembly abilities as identified by 1H NMR, we were interested in their bulk self-assembly behaviour. The differential scanning calorimetry (DSC) of Llipo and Lamphi showed only one peak for the first-order phase transition, and polarized optical microscopy (POM) showed that no liquid crystal state existed. Lamphi had a slightly higher Tm (179 °C) than Llipo (166 °C) (Fig. S4†). After thermal annealing by DSC, Llipo and Lamphi were used to conduct small angle X-ray scattering (SAXS) experiments. For Llipo, a strong scatter peak was observed at q1 = 1.36 nm−1, and weak signals of 2q1 and 3q1 could also be observed, which correspond to a repeating length of d = 4.63 nm (Fig. 5a). On the other hand, diffraction peaks were observed at a smaller angle (q1 = 0.59 nm−1, 2q1 = 1.18 nm−1) for Lamphi (Fig. 5b), indicating longer repeating units of d = 10.6 nm. In MM2 force field optimization, the molecular length of Llipo and Lamphi were both 6.4 nm. We can deduce that a layered periodic structure exists in both compounds. The d spacing of Llipo was close to the molecular length indicating a repeating unit of one single molecule (Fig. 6, left), while the d spacing of Lamphi was almost twice as long as the molecular length corresponding to the repeating unit of two molecules, which indicates that phase separation of TEG and alkyl chains occur in Lamphi (Fig. 6, right). The observed length was smaller than the real value because of the overlapping of the alkyl chains. These results demonstrate that incompatible side chains assist D–A dyads to achieve donor–acceptor phase separation. For Slipo and Samphi, the DSC showed no phase transition before they were decomposed, so thermal treatment is not ideal for the annealing process (see DSC curves in Fig. S5†). Thus, solvent annealing was adopted. The dyad Samphi formed a gel during the slow solvent evaporation of its solution in a dichloromethane (DCM)–hexane (v/v = 1/1) mixture, and the Slipo solution remained clear under the same conditions (Fig. S6†). After evaporating the residual solvent under vacuum, the solid (Slipo) and dried gel (Samphi) were used to conduct a SAXS experiment. One scattering peak was obtained for each dyad (q = 1.40 nm−1 for Slipo, q = 0.83 nm−1 for Samphi). The smaller q value for Samphi indicated similar phase separation behaviour with long-linker dyads. However, the absence of high-order scattering demonstrated poor phase separation results or improper assembly conditions (Fig. 5c and d).
 | ||
| Fig. 5 Small angle X-ray scattering of Llipo (a), Lamphi (b), Slipo (c), Samphi (d) samples. Llipo and Lamphi were treated by DSC, and Slipo and Samphi were treated by solvent annealing. | ||
 | ||
| Fig. 6 Schematic phase separation diagram of long-linker dyads; Llipo has a multilamellar structure (left) and Lamphi has a bilayer structure (right). | ||
According to scanning electron microscopy, Slipo, Samphi and Lamphi form fibres while Llipo formed nanoparticles after a heating–cooling cycle in chloroform–MeOH (v/v = 1/1) at a concentration of 6 × 10−4 M (Fig. S14†).
To explore the assembly in detail, the concentration was lowered to 6 × 10−5 M. After heating and then cooling, the assemblies were observed by transmission electron microscopy (Fig. 7). In an alcohol-containing solvent, amphiphilic dyads can form a more regular nanofibre structure. Samphi formed thin fibres with a width of around 10 nm, which is about twice the length of the rigid core (3.5 nm). It's in agreement with the model proposed in the SAXS experiment. In contrast, without the regulation of side chains, Slipo could only form thick fibres. However, Llipo, which differed only in linker length from the Slipo dyad, formed nanoparticles with a diameter around 50 nm. For Lamphi with a longer molecular length than Samphi, nanofibres narrower than 10 nm were observed. We speculate the reasons for different morphology formation are as follows. At least two factors contribute to the morphology differences: (1) the properties of the side chains (amphiphilic or lipophilic); (2) the linker length of the dyads (long-linker ones can fold as evidenced by UV-vis absorption spectra, thus are more flexible than short-linker ones). The incompatible side chains enable amphiphilic dyads to form thin fibres. The thinner fibres for Lamphi may be attributed to the flexible long linker. Due to the lipophilic affinity of side chains and relative rigid molecular structure, Slipo formed thick fibres with a diameter larger than 50 nm. For Llipo, the flexible long linker allows the D–A molecules to fold, and the side-by-side intermolecular lipophilic interactions were disrupted, resulting in discrete nanoparticles.
 | ||
| Fig. 7 Transmission electron microscopy images of four dyads. | ||
Cyclic voltammetry
To verify whether the energy levels of the donor and acceptor match, cyclic voltammetry was carried out. The data are summarized in Table 1 (see the redox curve in Fig. S3†). The HOMO of T2BTZ and LUMO of PDI were determined by the onset of the CV curve relative to ferrocene. Their corresponding LUMO or HOMO was calculated from the optical bandgap obtained from the UV-vis absorption spectra. The short-linker dyads had HOMO and LUMO values nearly identical to T2BTZ and PDI, respectively, which also demonstrated the independent D–A electronic properties in the dyads. The LUMO difference between the donor and acceptor was 0.62 eV (Table 1), which was sufficient to provide a driving force for charge separation. The intramolecular quenching of T2BTZ fluorescence by PDI also indicates the matched energy levels (Fig. S2†). Therefore, reasonable energy levels enable the dyads to be promising materials in organic photovoltaics.Conclusions
Lipophilic and amphiphilic dyads were synthesized and unambiguously characterized. Their self-assembly behaviour was studied from dilute solutions to the bulk state.In chloroform, no obvious difference in assembly behaviour between amphiphilic and lipophilic dyads was observed, probably due to the good solubility of these dyads. In dilute solutions (10−6 to 10−5 M), only long-linker dyads showed intramolecular D–A interactions. However, in concentrated solutions (10−3 to 10−2 M), all four dyads showed D–A interactions and A–A aggregation. Among them, short-linker dyads showed stronger intermolecular interactions, evidenced by 1H NMR, maybe because the short-linker enhanced the intermolecular cooperativity of the D–A interaction and A–A aggregation.
Different from its assembly behaviour in solution, in the bulk state, with the assistance of amphiphilic chains, Lamphi can overcome D–A interactions and achieve better nanoscale D–A phase separation compared with Llipo. In contrast, Samphi has poor phase separation but good crystallinity. Therefore, Lamphi is more suitable for film formation processes. Under the same aggregate growth conditions, the assembly morphology of the four dyads is as follows: both of the amphiphilic dyads could form thin nanofibres (Φ ≤ 10 nm), while the lipophilic dyads form thick fibres or nanoparticles (Φ ≈ 50 nm). The phase separation and morphology results provide guidance for organic device fabrication. Photovoltaic device performance of the dyads is under investigation in our lab.
Experimental
Materials
All the chemicals were purchased commercially. The oxygen and moisture sensitive reactions were performed under a nitrogen atmosphere using standard Schlenk techniques. Tetrahydrofuran (THF) was freshly distilled with sodium under a nitrogen atmosphere prior to use. N,N-Dimethylformamide (DMF) was stirred overnight with CaH2, distilled under vacuum and then dried over molecular sieves.Instrumentation
1H and 13C NMR spectra were collected on a Mercury plus 300 (300 MHz) or Bruker Avance 400 (400 MHz) spectrometer, using CDCl3 or d6-DMSO as solvents. Chemical shifts are reported in parts per million (ppm) and coupling constants are reported in Hertz (Hz). 1H NMR chemical shifts were referenced to the residual solvent peak (7.26 ppm in CDCl3 or 2.50 ppm in d6-DMSO). 13C NMR chemical shifts were referenced to CDCl3 (77.0 ppm). Electron ionization (EI) mass spectra were collected on a VG ZAB-HS mass spectrometer. Elemental analyses were performed using a German Vario EL III elemental analyzer. Electro-spray ionization (ESI) mass spectrometry was performed using a Bruker Apex IV FTMS instrument. Matrix-Assisted Laser Desorption/Ionization Time of Flight Mass Spectrometry (MALDI-TOF-MS) was performed using an ABI 4800 Plus MALDI TOF/TOF™ Analyzer. The UV-vis absorption spectra were recorded using a Hitachi U-4100 spectrophotometer with a 1 cm quartz cell. The photoluminescence spectra were recorded on a Horiba Jobin Yvon FluoroMax-4P spectrofluorometer with right-angle geometry, using a 1 cm quartz cuvette. Cyclic voltammetry was carried out by using BASI Epsilon workstation. The compounds were detected in DCM (freshly distilled over CaH2 prior to use) containing 0.1 M n-Bu4NPF6 as an electrolyte. The working electrode was a glassy carbon electrode and the counter electrode was a platinum sheet electrode. The redox potentials were recorded versus a Ag/AgCl electrode (reference electrode). Differential scanning calorimetry was carried out on a METTLER TOLEDO DSC 822 with a programmed heating procedure under nitrogen. Small-angle X-ray scattering experiments were carried out on a SAXSess high-flux small-angle X-ray scattering instrument (Anton Paar), equipped with a Kratky block-collimation system, at room temperature. The X-ray was generated by a sealed-tube X-ray generator (Philips PW3830) with a Cu target (λ: 0.1542 nm). The power of the generator used for measurement was 40 kV and 40 mA. The X-ray intensities were recorded on an imaging-plate detection system with a pixel size of 42.3 × 42.3 μm2. The distance from the sample to the detector was 264.5 mm and the exposure time was 600 s. The Scanning Electron Microscopy (SEM) experiment was carried out on a Hitachi S4800 Cold Field Emission Scanning Microscope. Transmission electron microscopy was carried out on a JEOL JEM-2100F field-emission high resolution transmission electron microscope.Synthesis
The p-type (Scheme 1) and n-type (Scheme 2) segments were synthesized separately and then connected with amino acid linkers (Scheme 3). Synthetic details of all compounds were summarized in the ESI.†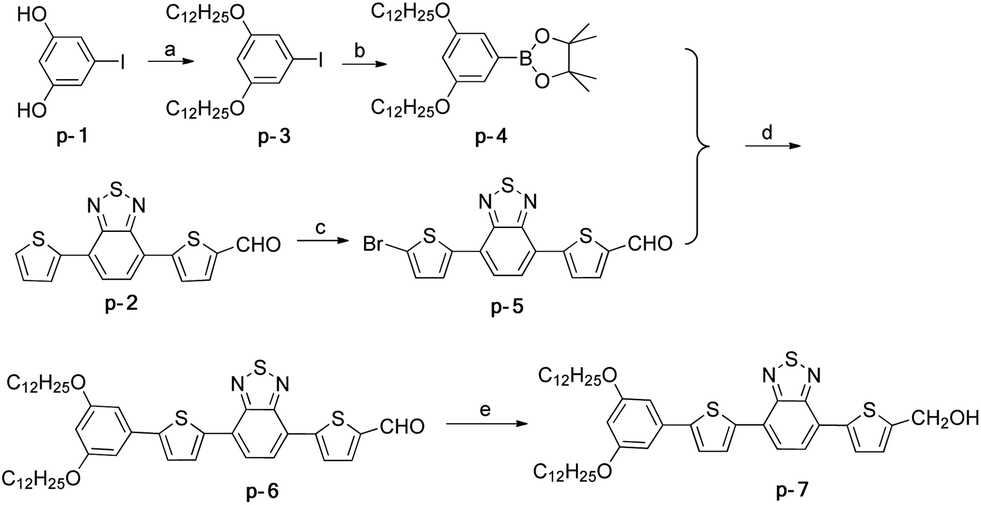 | ||
| Scheme 1 Reagents and conditions for the synthesis of the p-type segment: (a) n-C12H25Br, K2CO3, KI, acetone, reflux, 83%; (b) bis(pinacolato)diboron, PdCl2(dppf), KOAc, DMF, 60 °C, 74%; (c) N-bromosuccinimide, CHCl3–AcOH, 73%; (d) Pd(PPh3)4, K2CO3 (2 M), THF, 80 °C, 76%; (e) LiAlH4, THF, 89%. | ||
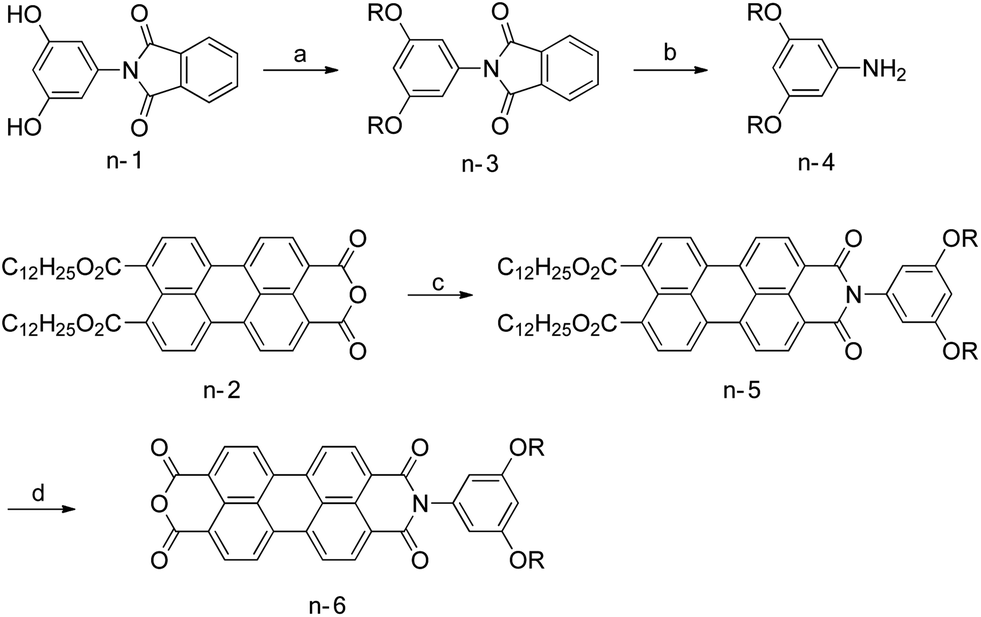 | ||
| Scheme 2 Reagents and conditions for the synthesis of the n-type segments: R = R1, (a) n-C12H25Br, K2CO3, DMF, 80 °C, 91%; (b) N2H4·H2O, EtOH–THF, reflux, 82%; (c) n-4, 4-dimethylaminopyridine, imidazole, 130 °C, 79%; (d) p-toluenesulfonic acid monohydrate, 95 °C, toluene, 88%. R = R2, (a) p-toluenesulfonyl triethylene glycol monomethyl ether, K2CO3, DMF, 90 °C, 47%; (b) N2H4·H2O, EtOH, reflux, 91%; (c) n-4, 4-dimethylaminopyridine, imidazole, 130 °C, 75%; (d) p-toluenesulfonic acid monohydrate, 95 °C, toluene, 88%. | ||
 | ||
| Scheme 3 Reagents and conditions for the synthesis of the four dyads: R = R1, (a) imidazole, DMF, 95 °C, H2N(CH2)5CO2H, 89%; (b) p-7, diisopropyl azodicarboxylate, PPh3, THF, rt, 30 min, 54%; (c) imidazole, DMF, 95 °C, glycine, 92%; (d) (COCl)2, DMF, DCM, rt, 2 h; 4-nitrophenol, Et3N, DCM, overnight, 91%; (e) p-7, 4-dimethylaminopyridine, DMF, 72 h, 53%. R = R2, (a) imidazole, DMF, 95 °C, H2N(CH2)5CO2H, 89%; (b) p-7, Et3N, DCM, 2-chloro-1-methylpyridinium iodide (Mukaiyama's salt), 34%. (c) Imidazole, DMF, 95 °C, glycine, 62%; (d) (COCl)2, DMF, DCM, rt, 2 h; 4-nitrophenol, Et3N, DCM, overnight, 47%; (e) p-7, DCM, DMF, 72 h, 69%. | ||
p-7 was synthesized from p-1 and p-2 in 5 steps. The diphenol p-1 and aldehyde p-2 were prepared according to the literature,13 and then p-1 was treated through alkylation and Miyaura boration to afford p-4. p-6 was obtained via a Suzuki coupling of p-4 and p-5. The reduction of p-6 by LiAlH4 gave the final p-7(Scheme 1).
n-7 and n-8 were afforded by the unsymmetrical functionalization of perylene diimide. Starting materials n-1 and n-2 were synthesized according to the literature.14 Alkylation of n-1 followed by Gabriel synthesis afforded the aniline derivative n-4, which can be condensed with anhydride n-2. The perylene monoimide diester was treated with p-toluenesulfonic acid to afford another anhydride, n-6. n-6 was condensed with 6-aminocaproic acid or glycine to obtain n-7 or n-8, respectively.
Various condensation methods were tested in order to link both segments. The Mitsunobu reaction and Mukaiyama condensation were finally applied for Llipo and Lamphi, respectively (Scheme 3). Carboxylic acid n-8 has lower condensation reactivity than n-7. The possible reasons were proposed as follows: (1) n-8 bearing a glycine linker has extremely poor solubility in common solvents (THF, CHCl3, DCM, toluene, etc.); (2) the hydroxyl group of n-8 may be deactivated by forming intramolecular hydrogen bonds with one of the carbonyl oxygens of a diimide unit. To overcome these disadvantages, we utilised transesterification.15 The active ester n-9, prepared from acid chloride, has an enhanced solubility, which allows a mild conversion to obtain the target compounds Slipo and Samphi (Scheme 3).
Acknowledgements
This research was financially supported by National Natural Science Foundation (no. 21074004, 91227202), the Ministry of Science and Technology (no. 2013CB933501), and the Ministry of Education (NCET-08-0005) of China.Notes and references
- (a) S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324 CrossRef PubMed; (b) W. Ma, A. Gopinathan and A. J. Heeger, Adv. Mater., 2007, 19, 3656 CrossRef CAS; (c) H. Hoppe and N. S. Sariciftci, J. Mater. Chem., 2006, 16, 45 RSC.
- (a) D. R. Kozub, K. Vakhshouri, S. V. Kesava, C. Wang, A. Hexemerb and E. D. Gomez, Chem. Commun., 2012, 48, 5859 RSC; (b) P. E. Shaw, A. Ruseckas and I. D. W. Samuel, Adv. Mater., 2008, 20, 3516 CrossRef CAS.
- G. Yu, J. Gao, G. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS.
- (a) Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761 CrossRef CAS PubMed; (b) Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, N. Ishii and T. Aida, J. Am. Chem. Soc., 2007, 129, 9276 CrossRef CAS PubMed; (c) P. Samorì, X. Yin, N. Tchebotareva, Z. Wang, T. Pakula, F. Jäckel, M. D. Watson, A. Venturini, K. Müllen and J. P. Rabe, J. Am. Chem. Soc., 2004, 126, 3567 CrossRef PubMed; (d) C.-L. Wang, W.-B. Zhang, C.-H. Hsu, H.-J. Sun, R. M. Van Horn, Y. Tu, D. V. Anokhin, D. A. Ivanov and S. Z. D. Cheng, Soft Matter, 2011, 7, 6135 RSC; (e) T. N. Y Hoang, D. Pociecha, M. Salamonczyk, E. Gorecka and R. Deschenaux, Soft Matter, 2011, 7, 4948 RSC.
- (a) W.-S. Li, Y. Yamamoto, T. Fukushima, A. Saeki, S. Seki, S. Tagawa, H. Masunaga, S. Sasaki, M. Takata and T. Aida, J. Am. Chem. Soc., 2008, 130, 8886 CrossRef CAS PubMed; (b) W.-S. Li, A. Saeki, Y. Yamamoto, T. Fukushima, S. Seki, N. Ishii, K. Kato, M. Takata and T. Aida, Chem. – Asian J., 2010, 5, 1566 CrossRef CAS PubMed; (c) R. Charvet, Y. Yamamoto, T. Sasaki, J. Kim, K. Kato, M. Takata, A. Saeki, S. Seki and T. Aida, J. Am. Chem. Soc., 2012, 134, 2524 CrossRef CAS PubMed; (d) F. Würthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C.-C. You, P. Jonkheijm, J. v. Herrikhuyzen, A. P. H. J. Schenning, P. P. A. M. van der Schoot, E. W. Meijer, E. H. A. Beckers, S. C. J. Meskers and R. A. J. Janssen, J. Am. Chem. Soc., 2004, 126, 10611 CrossRef PubMed.
- (a) M. E. El-Khouly, A. M. Gutiérrez, Á. Sastre-Santos, F. Fernández-Lázaro and S. Fukuzumi, Phys. Chem. Chem. Phys., 2012, 14, 3612 RSC; (b) T. K. Khan and M. Ravikanth, Tetrahedron, 2012, 68, 830 CrossRef CAS PubMed; (c) P. Piotrowiak, Chem. Soc. Rev., 1999, 28, 143 RSC; (d) F. D'Souza and O. Ito, Chem. Commun., 2009, 4913 RSC; (e) G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Chem. Rev., 2010, 110, 6768 CrossRef CAS PubMed.
- (a) D. Markovitsi, H. Bengs and H. Ringsdorf, J. Chem. Soc., Faraday Trans., 1992, 88, 1275 RSC; (b) E. H. A. Beckers, S. C. J. Meskers, A. P. H. J. Schenning, Z. Chen, F. Würthner, P. Marsal, D. Beljonne, J. Cornil and R. A. J. Janssen, J. Am. Chem. Soc., 2006, 128, 649 CrossRef CAS PubMed; (c) W. Pisula, M. Kastler, D. Wasserfallen, J. W. F. Robertson, F. Nolde, C. Kohl and K. Müllen, Angew. Chem., Int. Ed., 2006, 45, 819 CrossRef CAS PubMed.
- J.-L. Wang, Q. Xiao and J. Pei, Org. Lett., 2010, 12, 4164 CrossRef CAS PubMed.
- M. Ince, M. V. Martínez-Díaz, J. Barberá and T. Torres, J. Mater. Chem., 2011, 21, 1531 RSC.
- W. Wang, L.-S. Li, G. Helms, H.-H. Zhou and A. D. Q. Li, J. Am. Chem. Soc., 2003, 125, 1120 CrossRef CAS PubMed.
- (a) A. M. Ballantyne, T. A. M. Ferenczi, M. Campoy-Quiles, T. M. Clarke, A. Maurano, K. H. Wong, W. M. Zhang, N. Stingelin-Stutzmann, J. S. Kim, D. D. C. Bradley, J. R. Durrant, I. McCulloch, M. Heeney, J. Nelson, S. Tierney, W. Duffy, C. Mueller and P. Smith, Macromolecules, 2010, 43, 1169 CrossRef CAS; (b) M. M. Ling and Z. Bao, Chem. Mater., 2004, 16, 4824 CrossRef CAS.
- Y.-J. Cheng, S.-H. Yang and C.-S. Hsu, Chem. Rev., 2009, 109, 5868 CrossRef CAS PubMed.
- (a) W.-w. Mao, T.-t. Wang, H.-p. Zeng, Z.-y. Wang, J.-p. Chen and J.-g. Shen, Bioorg. Med. Chem. Lett., 2009, 19, 4570 CrossRef CAS PubMed; (b) T.-W. Zeng, C.-C. Ho, Y.-C. Tu, G.-Y. Tu, L.-Y. Wang and W.-F. Su, Langmuir, 2011, 27, 15255 CrossRef CAS PubMed.
- (a) M. Peterca, M. R. Imam, C.-H. Ahn, V. S. K. Balagurusamy, D. A. Wilson, B. M. Rosen and V. Percec, J. Am. Chem. Soc., 2011, 133, 2311 CrossRef CAS PubMed; (b) A. Wicklein, M.-A. Muth and M. Thelakkat, J. Mater. Chem., 2010, 20, 8646 RSC.
- J. Hongrapipat, P. Kopečková, S. Prakongpan and J. Kopeček, Int. J. Pharm., 2008, 351, 259 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, details of spectroscopic and analytical data. See DOI: 10.1039/c3ra47633b |
| This journal is © The Royal Society of Chemistry 2014 |