Metal binding properties of pyrimidinophanes and their acyclic counterparts†
Sergey N. Podyachev*a,
Vyacheslav E. Semenova,
Victor V. Syakaeva,
Nadezda E. Kashapovab,
Svetlana N. Sudakovaa,
Julia K. Voroninaa,
Anatoly S. Mikhailova,
Alexandra D. Voloshinaa,
Vladimir S. Reznika and
Alexander I. Konovalova
aA. E. Arbuzov Institute of Organic and Physical Chemistry, Kazan Scientific Center of Russian Academy of Sciences, Arbuzov str., 8, 420088, Kazan, Russia. E-mail: spodyachev@iopc.ru; Fax: +7 843 2731872
bKazan National Research State Technological University, K. Marks str., 68, 420015, Kazan, Russia
First published on 30th January 2014
Abstract
A series of acyclic and macrocyclic nucleobase derivatives that contain uracil and 2-thiocytosine units linked by polymethylene spacers was prepared. The length of spacers as well as the oxidation number of sulfur atoms entering into the structure of the compounds was varied. The crystal structure of pyrimidinophane with hexa- and heptamethylene linkers was established by X-ray analysis. The metal ion-binding properties of pyrimidinophanes and their acyclic counterparts were investigated by liquid extraction and 1H NMR titration experiments. The compounds with 2-thiocytosine fragments revealed a high selectivity of extraction towards Ag+ ions. The stoichiometry of silver complexes and the binding constants have been determined. The antibacterial activity of these complexes was also discussed.
Introduction
Nitrogen-containing heterocycles and especially pyrimidine derivatives are widespread in nature and are essential for life, playing a vital role in the metabolism of all living cells.1 At the same time, metal ions play an important role in the function of nucleic acids. The transfer of genetic information,2 replication processes, transcription and translation as well as the conformational transitions3,4 proceed with the participation of metal ions. Moreover, metal ions can be involved in the catalytic action of ribozymes.5–8 The binding of metal ions by pyrimidine base pairs in DNA duplexes is also a subject of up-to-date investigations.9 Complexes of nucleobases with metal ions can be considered as attractive model systems for the analysis of the interaction of metal ions with nucleic acids. The studying of coordination compounds of nucleotide bases makes possible to identify the donor centers which are responsible for the complex formation and determine the structure of coordination polyhedral as well as the quantitative characteristics of complex formation process. In addition, the transition metal complexes with N-heterocycles (e.g., pyrimidine derivatives) are appropriate objects for supramolecular chemistry. Their possibility to form both the coordination and the hydrogen bonds makes them promising for the use in materials with supramolecular architecture.10–14There is much information about complexes of transition and noble metals with the nucleotide bases,15–17 especially uracil18 in the current literature. However, data on the complex formation between the metal ions and more sophisticated 3D ligands possessing an acyclic or cyclic topology and containing several nucleic acid fragments are virtually lacking. In particular, pyrimidinophanes are the cyclic receptors with one and more nucleobase derivatives.19 However, only the pyrimidinophanes with crown-ether bridges were reported to form complexes with some of alkali and alkaline-earth metals.20–22
Recently, we have synthesized a new class of pyrimidinophanes. In addition, the acyclic bispyrimidine as well as trispyrimidine capable of simulating the building fragments of these compounds were also reported.23–25 The synthesized pyrimidinophanes consist of the two 2-thiocytosine (4-amino-6-methyl-2-thiopyrimidinyl) and one 6-methyluracil fragments linked by the polymethylene spacers. We have earlier isolated and characterized complexes of Cu(II) with some of pyrimidinophanes and their acyclic counterparts.26 It should be noted that uracil as well as 5-methyluracil (thymine) moieties enter into the composition of the nucleic acids of RNA and DNA, and 2-thiocytosine is known as a modified nucleobase found in the tRNA of some organisms.27 It was also reported that 2-thiocytosine and its derivatives as well as their complexes possess antileukemic,28,29 anticancer,30,31 antitumor,32–34 antiviral35 and antimycobacterial36 activities.
In the present work, we describe the synthesis and coordination properties of some earlier and newly synthesized pyrimidinophanes and their acyclic building blocks (Scheme 1). To evaluate the binding abilities of the synthesized compounds towards the metal ions, we have performed the investigation of their complex formation properties, using the liquid extraction and NMR methods. We have also studied the complexation of simple pyrimidine derivatives 1 and 2 to establish the influence of the topology of compounds (podands 3, 4 and macrocycles 7a–d) on their coordination properties. Moreover, in order to identify the role of sulfur donor atoms in the coordination of metal ions we have synthesized some novel pyrimidinophanes by the oxidation of sulfenyl (>S) moieties of the macrocyclic and acyclic compounds to the sulfinyl (>SO) (compound 5) and sulfonyl (>SO2) (compounds 6 and 8) ones. Afterwards, the binding properties of the synthesized compounds have been explored and compared.
 | ||
| Scheme 1 The structural formulae of investigated compounds 1–8. | ||
Results and discussion
Synthesis of the pyrimidinophanes and their acyclic counterparts
New pyrimidinophanes 7a,c were prepared by the reaction of disodium salts of N,N′-(alkane-α,ω-diyl)bis(4-amino-6-methylpyrimidine-2-thiones) 9a,b with dibromide 10 in DMF (Scheme 2). The macrocycles were obtained with the yields of 22% and 14%, respectively. This approach for the synthesis of pyrimidinophanes having the similar structure was developed by us previously.23 | ||
| Scheme 2 Synthesis of pyrimidinophanes comprising two thiocytosine and one uracil moieties. Reagents and conditions: (i) DMF, room temperature, 30 h. | ||
To elucidate donor sites of the pyrimidinophanes and their acyclic counterparts participating in the coordination with metal ions, the sulfinyl and sulfonyl groups were introduced into the compounds by the oxidation of S atoms at the pyrimidine rings. Oxidation of thioalkyl substituents of the acyclic compound 4a with meta-chloroperoxybenzoic acid at −40 ÷ −45 °C afforded disulfoxide 5 with 71% yield. Apart from disulfoxide 5, the disulfone 6 was also isolated from the reaction mixture with 9% yield. The macrocyclic trispyrimidine 7e, on the contrary, wasn't oxidized under such conditions. It should be also noted that this pyrimidinophane was oxidized with the formation of macrocyclic disulfone 8 (86%) at the room temperature. It seems that the forming of oxidation products for the trispyrimidines is determined by the topology (acyclic or macrocyclic) of starting compounds.
The compounds were characterized by the combination of NMR, IR and MS techniques. The spatial structure of 7c was finally established by single crystal X-ray crystallography. As shown in Fig. 1a, the pyrimidinophane possesses an expanded structure formed due to the stretched hexa- and heptamethylene chains. The 2-thiocytosine fragments are slightly twisted relative to each other (dihedral angle P1/P2 is equal 16.32(3)°), at the same time the uracil unit is turned towards the both rings in a greater extent. Thus, the dihedral angles P1/P3 and P2/P3 have the magnitudes 56.94(3)° and 40.64(3)°, respectively. Each molecule is linked with a pair of adjacent molecules via classical H-bonds formed by two NH groups, carbonyl oxygen atom of the uracil unit and one of the sulfur atoms. The turn of uracil fragment and the intermolecular hydrogen bonds lead to the formation of zig-zag pyrimidinophane ribbons due to the participation of each molecule in donor and acceptor interactions (Fig. 1b).
 | ||
| Fig. 1 Classical intermolecular H-bonds (N(20)–H(20)⋯O(43): N(20)⋯O(43) 2.864(8) Å, N(20)–H(20)⋯O(43) 164°; N(28)–H(28)⋯S(13): N(28)⋯S(13) 3.631(6) Å, N(28)–H(28)⋯S(13) 166°) (a) and supramolecular ribbons (b) in the crystal of 7c. The labels P1–P3 indicate the structural fragments containing pyrimidine (C14–C19 (P1), (C29–C34) (P2)) and uracil (N1–C6 (P3)) rings of pyrimidinophane. | ||
Extraction studies
Experiments on liquid extraction of s- (Li+, Na+, K+, Cs+ and Ca2+), p- (Pb2+), d- (Co2+, Ni2+, Cu2+, Zn2+, Ag+, Cd2+ and Hg2+) and f- (La3+, Gd3+ and Lu3+) elements were accomplished in two-phase (water–chloroform) system, using the picrate method.37,38 The presence of heteroatoms with lone electron pairs in the investigated ligands provides their basic properties. Protonation of these ligands (L) in acidic conditions leads to the formation of an ion-pair complex with the picrate anion (Pic−) and, as a consequence, to the transfer of picric acid from aqueous phase to the organic, even in the absence of metal ions in the aqueous layer (eqn (1), Fig. 2).| zHPicaq + nLorg ⇆ [LnH+z(Pic−)z]org | (1) |
 | ||
| Fig. 2 Effect of pH on a degree of transfer of HPic in the systems containing extractants 1–8 ([HPic] = 2.5 × 10−4 M; [L1] = 2 × 10−3 M; [L2–8] = 1 × 10−3 M). | ||
The transfer of picric acid in the presence of the model compound 2-thiomethyl-4-dimethylamino-6-methylpyrimidine 1 is rather noticeable even at pH ≤ 6 (E > 25%), while the same process in the case of 1,3,6-trimetiluracil 2 is observed only at pH ≤ 2.5 (E > 25%). Obviously, the main contribution to the transfer process for the compound 1 belongs to the nitrogen and/or sulfur atoms of the 2-thiocytosine fragment.
The incorporation of two pyrimidine fragments into the structure of podand (compound 4b) and pyrimidinophane (compounds 7a–d) only slightly effects on the transfer of picric acid. The attachment of oxygen atoms to the sulfur atoms, when going from podand 4b to podands 5 and 6, and especially, from pyrimidinophane 7e to pyrimidinophane 8, leads to a drastic decrease of the transfer of picric acid. Apparently, the main reason explaining this behavior of compounds is the weakening of the basicity properties of the nitrogen atoms of a pyrimidine ring caused by the influence of electron withdrawing sulfoxide or sulfone groups. It is interesting to note that the closure of podand 6 into the macrocyclic structure 8 also results in the significant decrease of the picric acid transfer from the aqueous to the organic layer, which is probably associated with the steric hindrances in the process of salt formation.
The increase of pH in the aqueous layer prevents the protonation of the investigated compounds. Therefore, for the correct comparison of extraction data and the determination of stoichiometry of the complexes formed during extraction the experiments were carried out at pH = 7.3, supported by the buffer. In such conditions the transfer of picric acid from the aqueous to the organic phase did not exceed 8% (Fig. 2). It should be noted that the experiments accomplished at higher pH lead to the undesirable hydrolysis of metal ions. Thus, our attempts to adjust pH of the solutions of mercury(II) nitrate to the value 7.3 unfortunately resulted in the precipitation of mercury(II) oxide. Therefore, the investigations of the extraction properties of synthesized compounds 1–8 with Hg2+ and Pb2+ ions have not been performed. The process of Ag+ recovery by the pyrimidinophanes 7b and 7d was also accompanied by the precipitation. Therefore, we could not accomplish the further investigation of the extraction properties of these ligands.
If to ignore the insignificant transfer of picric acid, the extraction process can be described by the eqn (2):
| Mz+aq + zPic−aq + nLorg ⇆ [Mz+LnPic−z]org, | (2) |
Extraction data for the metal picrates recoverable by pyrimidine derivatives 1–8 are presented in Fig. 3. The figure shows that alkali (Li+, Na+, K+, Cs+) and alkaline earth (Ca2+) metal ions are not practically extracted by compounds 1–8. Some extraction efficiency towards these metal ions, revealed by the compounds 6 and 7c is obviously caused by the transfer of picric acid (Fig. 2). The percentage of extraction of lanthanide ions, particularly Gd3+ and Lu3+, is notably higher as compared to similar data for alkali metal ions, indicating a quite more efficient binding ability of the former. Cu2+ and Ag+ ions are bound more effectively than the other d-ions. However, the most effective extraction of Cu2+ achieved by the compound 7c did not exceed 19% (Fig. 3). In the same conditions, Ag+ ions are extracted by compounds 3–5 and 7a,c almost quantitatively (99–100%). 2-Thiomethyl-4-dimethylamino-6-methylpyrimidine 1 binds effectively Ag+ ions (∼95%) and insignificantly Cu2+ ions (∼8%). Thus, all the investigated compounds, excepting the uracil derivative 2, selectively bind Ag+ ions.
 | ||
| Fig. 3 Extraction percentages (E%) of metal picrates from water into CHCl3 at 25 °C by 1–3, 4b, 5, 6, 7a,c, 8. (C1 = 2 × 10−3 M; C3,4b,5,6,7a,7c,8 = 1 × 10−3 M; CHPic = 2.5 × 10−4 M; CMz+ = 1 × 10−2 M; pH = 7.3). | ||
According to the concept of hard and soft acids and bases (HSAB), the introduction of soft donor atoms of nitrogen and especially sulfur atoms into the molecules should improve their binding ability towards soft metal ions. From the other hand, the preorganization of binding groups on the macrocyclic platform can be resulted in an additional enhancement of their affinity towards various substrates caused by a cooperative effect of these groups. Indeed, the macrocyclic compounds based on calix[4]arenes functionalized by thioether,39 thiocrown ethers,40 thioamide41 or thiophosphate42 groups demonstrate a high extraction efficiency and selectivity for Ag+. Thus, a quantitative extraction of Ag+ (E = 99%) was observed for the p-tert-butyltetrathiacalix[4]arene substituted at the narrow rim by diethylthiophosphate ester groups (–PS(OEt)).43 However, some of metal ions have been also notably extracted by this compound (for example, E(Cu2+) = 11%). Reviews44–48 have comprehensively summarized the extraction data obtained for the liquid–liquid extraction of metal ions by podands, crown ethers and calixarenes. Among these compounds the 5,11,17,23-tert-butyl-25,26,27,28-(2-N,N-dimethyldithiocarbamoylethoxy)-calix[4]arene has exhibited the highest selectivity towards Ag+ (S = 18) relatively Cd2+ and Pb2+ ions.49 Unfortunately, the selectivity with respect to other usually efficiently extracted d-ions, such as Cu2+, was not studied by the authors. We have previously investigated the tetrathiacalix[4]arenes functionalized by tetraphenylhydrazone groups in the same range of metal ions.50 The extraction selectivity of Ag+ reached the similar value (S = 18). In the case of podand 5 and pyrimidinophane 7a, the selective extraction of Ag+ in the row of the investigated metal ions is much higher than for mentioned above compounds: S = E(Ag+)/E(Mz+) ≈ 50.
When the concentration of extractant was decreased by four times down to the equimolar ratio (Ag+Pic−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :L = 1:1), the efficiency of Ag+ recovery by compound 3 remained practically unchanged, whereas for other compounds it was markedly reduced. This fact can indicate a high affinity of podand 3 towards Ag+. The investigated pyrimidines according to their Ag+ extraction efficiency can be ranged in the following order (Fig. 4):
:L = 1:1), the efficiency of Ag+ recovery by compound 3 remained practically unchanged, whereas for other compounds it was markedly reduced. This fact can indicate a high affinity of podand 3 towards Ag+. The investigated pyrimidines according to their Ag+ extraction efficiency can be ranged in the following order (Fig. 4):
 | ||
| Fig. 4 Extraction percentages (E%) of silver picrate from water into CHCl3 at 25 °C by 1–3, 4b, 5, 6, 7a,c, 8. pH = 7.3; [HPic] = 2.5 × 10−4 M, [Ag+] = 1 × 10−2 M, [L1] = 5 × 10−4 M, [L3,4b,5,6,7a,7c,8] = 2.5 × 10−4 M. | ||
2 < 8 < 6 < 1 < 5 < 7c ≈ 7a < 4b < 3.
The integration of two 2-thiocytosine fragments through the hexamethylene spacer –(CH2)6– into the structure of compound 3 having the chelate configuration seems to be rather preferable for the cooperative coordination. In the case of podand 4b, wherein 2-thioalkyl-4-dimethylamino-6-methylpyrimidine fragments are attached to the 6-methyluracyl moiety by the hexamethylene chains, the binding efficiency revealed by this compound is practically the same. The presence of bulky substituents close to the sulfur and adjacent nitrogen atoms may prevent the effective coordination of metal ion. On the other hand, the increase of lipophilic properties of the ligand 4b in comparison with 3 should facilitate more effective transfer of the metal ion from aqueous to the organic phase.
It could be expected that the replacement of the hexamethylene chains by the tetramethylene ones in compound 5 compared to 4b should be resulted in the enhancement of the cooperative binding of a metal ion by two 2-thiocytosine fragments. However, the extraction efficiency for Ag+ in the case of the compound 5 is notably less (Fig. 4). Apparently, the replacement of sulfenyl group (compound 4b) by the sulfinyl one (podand 5) eliminates the cooperative contribution. The introduction of sulfonyl groups leads to a drastic decrease in the extraction efficiency (podand 6 and pyrimidinophane 8). It may be explained by the “exclusion” of sulfur atoms from the coordination with metal ion or by the weakening of the basicity of nitrogen atoms in the pyrimidine ring because of the electron withdrawing influence of SO2-group.
The extremely high binding efficiency of pyrimidines towards Ag+ ions makes difficult the correct estimation of the composition and extraction constants of the complexes from the dependence of extraction efficiency on a free ligand concentration due to the large experimental errors. Therefore, the stoichiometry of the extracted complexes with Ag+ was determined by using Job's method, when the total concentration of extractant and the metal picrate under experiment was constant (Fig. 5).
 | ||
| Fig. 5 Job's plots for Ag+Pic− extracted by 1, 3, 4b, 5, 7a and 7c. [HPic]aq + [L]org = 2.5 × 10−4 M, [Mz+] = 1 × 10−2 M, pH = 7.3. | ||
The transfer of Ag+Pic− from the aqueous to organic layer reached a maximum at 0.33 mole fraction in the case of compound 1, and for the podands 3, 4b, 5 as well as pyrimidinophanes 7a,c, the maximum of metal picrate transfer was observed at 0.5 mole fraction of Ag+Pic−, which indicates the formation of 1:2 (Ag+·2L1·Pic−) and 1:1 (Ag+·L3,4b,5,7a,7c·Pic−) complexes, respectively.
Complexation studies
:2 (peak [Ag·2L]+ for L1) and 1:1 (peaks [Ag·L]+ for L3,4b,7a) stoichiometry. The picrate anion does not contribute to the signals of mass spectra. This fact is obviously provided by the partially degradation of these complexes, which can be caused by weak binding of the Pic− anion due to its participation in the outer sphere coordination.Since solubility of these complexes in nonpolar solvents is quite negligible, the IR spectra for them were recorded in KBr pellets. The assignment of the absorbance bands in IR spectra has been performed in accordance with the methodology developed for the acyclic and macrocyclic pyrimidine compounds25,51 and some of their complexes with CuBr2 (ref. 26) investigated by us previously. The analysis of IR spectra makes possible to highlight some common spectral peculiarities of the spectra for the compounds and their complexes. The characteristic bands of 2-thiocytosine fragments in the spectra of the ligands are shifted to higher frequencies for their Ag+ complexes (from 1585 cm−1 to 1602 cm−1 for 1, from 1594 cm−1 to 1606 cm−1 for 3, from 1584 cm−1 to 1603 cm−1 for 4b and from 1589 cm−1 to 1602 cm−1 for 7a). At the same time, the absorbance bands for the carbonyl group ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) in the spectra of complexes 4b and 7a, comprising uracil fragments, were not shifted at all (Δν(CO) ≤ 1 cm−1). The maxima of absorption bands ν(CH3(N)) for the complexes of 1 and 4b remained the same. On going to Ag+ complex, the band of absorbance ν(NH) of ligand 3 detected at 3243 cm−1 is shifted to 3270 cm−1 and a new band at 3359 cm−1 appears in its spectrum. These changes are obviously caused by the weakening of hydrogen bonds N–H⋯X (X = Npyrim, O, or S). In the case of pyrimidinophane 7a, the absorption band ν(NH) at 3350 cm−1 is found in the spectrum both of ligand and complex, while the bands at 3321 cm−1 and 3245 cm−1 coalesce into the band at 3279 cm−1 when going to Ag+ complex. This fact is obviously explained by the changes in the system of hydrogen bonds occurring in the complex. Unfortunately, the absorption bands of ν(C–S) which usually appear in the region of 600–700 cm−1 are difficult to identify because of a low intensity of these signals in the spectra of the complexes. Generally, IR spectra analysis of the complexes in solid indicates that coordination of Ag+ cations by cyclic and acyclic ligands 1, 3, 4b and 7a is realized through the ring-nitrogen atoms.
O) in the spectra of complexes 4b and 7a, comprising uracil fragments, were not shifted at all (Δν(CO) ≤ 1 cm−1). The maxima of absorption bands ν(CH3(N)) for the complexes of 1 and 4b remained the same. On going to Ag+ complex, the band of absorbance ν(NH) of ligand 3 detected at 3243 cm−1 is shifted to 3270 cm−1 and a new band at 3359 cm−1 appears in its spectrum. These changes are obviously caused by the weakening of hydrogen bonds N–H⋯X (X = Npyrim, O, or S). In the case of pyrimidinophane 7a, the absorption band ν(NH) at 3350 cm−1 is found in the spectrum both of ligand and complex, while the bands at 3321 cm−1 and 3245 cm−1 coalesce into the band at 3279 cm−1 when going to Ag+ complex. This fact is obviously explained by the changes in the system of hydrogen bonds occurring in the complex. Unfortunately, the absorption bands of ν(C–S) which usually appear in the region of 600–700 cm−1 are difficult to identify because of a low intensity of these signals in the spectra of the complexes. Generally, IR spectra analysis of the complexes in solid indicates that coordination of Ag+ cations by cyclic and acyclic ligands 1, 3, 4b and 7a is realized through the ring-nitrogen atoms.
NMR Ag+ cation complexation studies
The estimation of the ionophoric properties of pyrimidines accomplished by the picrate extraction method has demonstrated their high affinity towards Ag+ ions. To obtain further information on the cation binding properties of these derivatives in solution, specifically concerning the position of binding sites, the proton NMR titrations of podands 3, 4b and macrocycles 7a–c with silver picrate were performed in CDCl3–DMSO-d6 = 1:1 (v/v).
The titration of ligands 3, 4b and 7a–c with Ag+Pic− did not lead to the appearance of new signals, but caused the distinct changes in the chemical shifts, indicating a fast metal exchange between complexed and uncomplexed species in the NMR time scale at 303 K. During the titration of ligands, the change of the chemical shifts of the protons of uracil fragments as well as adjacent methylene groups was imperceptible (<0.02 ppm). The location of the signals of the aromatic protons of picrate anions in the spectra of compounds also did not change. At the same time, the distinct change in the chemical shift was observed in the region of the proton signals of 2-thiocytosine fragments and methylene groups neighbouring to them (Table S1†).
Fig. 6 shows the chemical shift changes for the selected protons of compound 4b plotted against the amount of Ag+Pic− added to the solution of CDCl3–DMSO-d6 or CDCl3–CD3OD (Δδ = δC − δL values denote the chemical shifts of the complex and free ligand respectively). The addition of Ag+ salt right up to 1.0 mol equivalent resulted in the increase of Δδ value which then became constant and independent on a further addition of the salt. The similar situation was observed for ligands 3 and 7a–c in CDCl3–DMSO-d6. These facts obviously indicate the direct participation of 2-thiocytosine fragments of the ligands in the binding of Ag+ ions followed by the formation of the complexes of 1:1 stoichiometry. The equal stoichiometry of the complexes with Ag+Pic− formed by the investigated compounds in NMR and extraction experiments is obviously caused by a similar nature of the coordination interactions between the ligands and metal ions in solutions.
 | ||
| Fig. 6 1H NMR titration of 4b with Ag+Pic− in (a) CDCl3–DMSO-d6 and (b) CDCl3–CD3OD. | ||
For ligands 3, 4b, 7a–c the largest values of Δδ were observed for NH (0.60 ÷ 0.83 ppm), Ar-H (0.16 ÷ 0.26 ppm) and Ar-CH3 (0.12 ÷ 0.3 ppm) protons of 2-thiocytosine fragments (Fig. 7). Apparently, the main contribution to the chemical shifts of protons of these groups is provided by the coordination of the one of nitrogen atoms in the 2-thiocytosine fragments with the metal ion. At the same time, nitrogen atoms of such groups as –N–(CH3)2 (Δδ = 0.08 ppm for complex 4b) and –N(H)–CH2– (Δδ(CH2) = 0.08 ÷ 0.14 ppm for complexes 3 and 7a,c), probably do not participate in this coordination. A high value of ΔδNH observed for 3 and 7a–c complexes is a consequence of the acidic nature of NH-group and may be also caused by its conjugation with 2-thiocytosine fragments. Additional information about the nitrogen atom involved in the coordination process could be certainly obtained from the analysis of Δδ15N values. Unfortunately, we have managed to receive the 15N experimental data only for N(1) atoms for free ligands 1, 3 and 4b as well as for their Ag+ complexes (Table S1†). Similar values of Δδ(N(1)) ≈ −31 ÷ −33 ppm determined from the spectra of these ligands can testify to the participation of the same nitrogen heteroatoms of the 2-thiocytosine fragments in the coordination with Ag+Pic−. The realization of such coordination pattern in the solutions evidently seems to be more preferable also. Taking these facts into account, it could be expected that N(1) atoms apparently participate in the complex formation of ligands 3 and 4b.
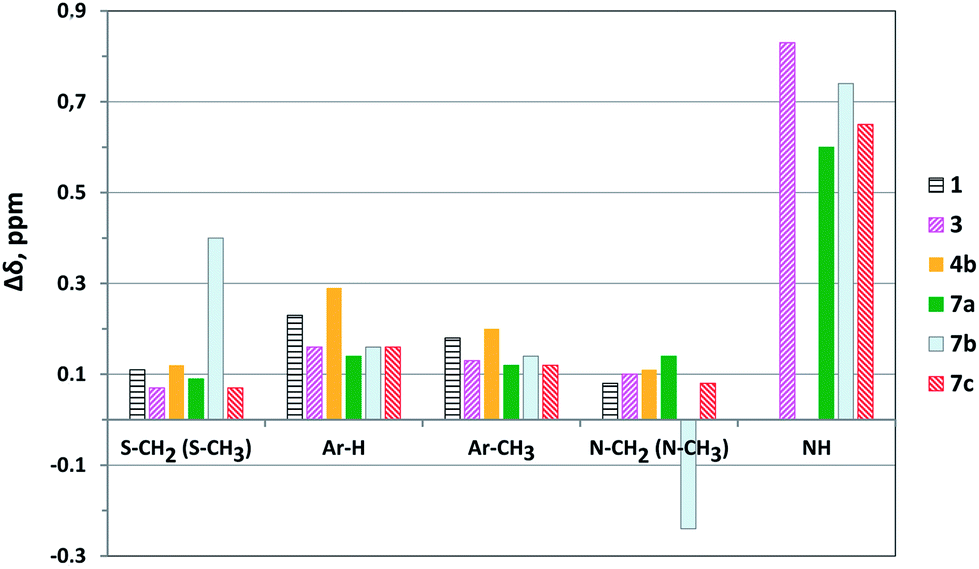 | ||
| Fig. 7 Chemical shifts' changes (Δδ, ppm) between Ag+ complexes and free ligands in CDCl3–DMSO-d6 = 1:1 (v/v) in 1H NMR at 303 K. | ||
The chemical shift changes for the protons of S–CH3 and S–CH2– groups in the spectra of the complexes for all investigated compounds are relatively small (Δδ = 0.07 ÷ 0.12 ppm). But only in the case of the pyrimidinophane 7b, the complex formation with Ag+ leads to the dramatically increased value of Δδ for the (S–CH2–) group of protons (Δδ(S–CH2–) = 0.4 ppm). Moreover, the abnormal change of the sign and value of ΔδN(H)–CH2– = −0.24 ppm for this compound is observed under going to Ag+ complex. It could be proposed for pyrimidinophane 7b (m = 5, n = 6) that the sulfur atoms obviously participate in the coordination with the metal ion, which may be resulted in a substantial transformation of its complex structure compared to 7a (m = 3, n = 6) and 7c (m = 7, n = 6) compounds. It is interesting to notice that the extraction of Ag+ by the macrocycle 7b in contrast to 7a and 7c having the tri- or heptamethylene spacers instead of the pentamethylene one was accompanied by the precipitation. This fact indirectly argues for the essential difference between the structures of the complexes formed.
The values of the stability constants (logβ) obtained from the analysis of Δδ(Ar-H(5)) for 2-thiocytosine fragments in the 1H NMR spectra of the investigated compounds are presented in the Table 1. The results obtained from the analysis of Δδ(NH) appeared to be almost similar. The stability constant of the 1:1 complex Ag+ with the ligand 3 has a value of logβ11 = 3.13 ± 0.03 in CDCl3–DMSO-d6. When going to pyrimidinophanes 7b (m = 5, n = 6) and 7c (m = 7, n= 6), the complex stability does not practically change (Table 1). At the same time, the decrease of the spacer length from the penta- to the trimethylene one leads to the noticeable decrease of the complex stability for pyrimidinophane 7a (m = 3, n = 6) (Δlogβ = logβ(Ag+ + 7b) − logβ(Ag+ + 7a) = 0.5 ± 0.2). Obviously, the length of the trimethylene spacer is not enough optimal for the realization of the effective coordination. A rather more noticeable binding is observed for the podand 4b in comparison with bispyrimidine 3 (Δlogβ = logβ(Ag+ + 4b) − logβ(Ag+ + 3) = 1.5 ± 0.2). The enhancement of the nitrogen atoms' basicity in 2-thiocytosine fragments after incorporation of electron donor –N(CH3)2 substitutes could be also considered as one of the reasons of this phenomenon.
β11) of Ag+ complexes in CDCl3–DMSO-d6 = 1:1 (v/v) at 303 K obtained by 1H NMR titration techniques
1H NMR titration of the podand 4b with the silver picrate was also accomplished in the solutions of CDCl3–CD3OD (1:1) (Fig. 6b). When going to a less polar solvent mixture, the significant downfield shifts (Table S1†) in the 1H NMR spectrum of 4b for its 2-thiocytosine fragments are observed (0.28–0.37 ppm). This fact probably indicates that a solution containing DMSO-d6 can solvate a ligand more effectively than CD3OD. The weakening of a concurrent interaction with a solvent promotes a high affinity of ligand 4b towards Ag+ ion (logβ > 5). Attempts to measure the stability constants for the Ag+ complexes of the compounds 3 and 7a–c in CDCl3–CD3OD failed due to the precipitation.
Thus, the investigated pyrimidinophanes as well as their acyclic counterparts form the stable 1:1 complexes with Ag+. The stabilization of Ag+ complexes is probably provided by the participation in the binding of both 2-thiocytosine fragments introduced into these compounds. It should be additionally noticed that a high selective binding of Ag+ by the simple pyrimidine bases (thymine, cytosine, 4-thiothymine, 2-thiothymine, 5-fluorouracil) in DNA duplexes has been observed earlier.9,52
Biological activity of the AgPic complexes with pyrimidine derivatives 1, 3, 4b and 7a
It is well known that silver ions are effective against a wide range of microorganisms and have a variety of antibacterial medical applications.53,54 The antimicrobial activity of silver ions is closely related to their interaction with nucleic acids, preferentially with the bases entering into the composition of DNA, as well as the thiol groups of proteins due to the binding of silver ions with key functional groups of enzymes.54 Keeping in mind the antimicrobial efficiency of silver ions and the biological activity of acyclic and macrocyclic 2-thiocytosine derivatives,23 it was interesting to determine the antimicrobial activity of silver complexes of the pyrimidinophanes and their acyclic counterparts.We have estimated in vitro the bacteriostatic and fungistatic activity of the AgPic and its complexes with ligands 1, 3, 4b, 7a against some of pathogenic representative Gram-negative and Gram-positive bacteria as well as the pathogenic fungi. Details of the biological evaluation are presented in ESI.† The AgPic showed a moderate activity against bacteria and fungi. The minimal bacteriostatic concentrations (MBC) of the salt against bacteria Staphylococcus aureus, Bacillus subtilis, Escherichia coli, in particular, had values of 3.9, 31.3 and 7.8 μg ml−1, respectively. The minimal fungistatic concentration (MFC) against yeast Candida albicans had a value of 31.3 μg ml−1. Coordination of AgPic with pyrimidine moieties of the ligands 1, 3, 4b and 7a (complexes Ag·2L1·Pic, Ag·L3·Pic, Ag·L4b·Pic, Ag·L7a·Pic) was followed by a dramatic increase of MBC and MFC values up to 31.3, 62.5 and 125 μg ml−1 towards bacteria Staphylococcus aureus, Bacillus subtilis, Escherichia coli, respectively, and 250 μg ml−1 towards the yeast (Table S2†). It is interesting to note that cytotoxic activity of the substituted 2-thiocytosines coordinated with organoplatinum compounds was found to be higher than that of platinum derivative itself, in particular, cisplatin.32–34
Conclusions
A series of acyclic and macrocyclic compounds with nucleobase derivatives contained uracil and 2-thiocytosine units linked by polymethylene spacers was prepared. The length of spacers as well as the oxidation number of sulfur atoms entering into the structure of the compounds was varied. According to RSA data, pyrimidinophane 7c with long hexa- and heptamethylene linkers in crystal possesses an expanded structure.The estimation of the extraction ability of these receptors in a wide range of metal ions by the liquid extraction method has demonstrated a high affinity of the investigated compounds towards Ag+ ion. The uniting of two 2-thiocytosine fragments in their structure was followed by the formation of the compounds with podand or macrocyclic topology and resulted not only in a significant increase of the efficiency, but also in a high selectivity of the extraction. Thus, the extraction selectivity for Ag+ relative to other studied metal ions in the case of bispyrimidine 5 as well as macrocycle 7b reached a value S = E(Ag+)/E(Mez+) ≈ 50, which exceeds the values reported before. The substitution of the sulfenyl or sulfinyl groups by the sulfonyl ones led to a drastic decrease in extraction efficiency for the synthesized compounds.
According to the liquid extraction and 1H NMR titration experiments, the investigated pyrimidinophanes as well as their acyclic counterparts form 1:1 complexes with Ag+ ions and have closely related stability constants (logβ = 2.8 ÷ 3.3), except for the bispyrimidine 4b comprising two 2-thiocytosine fragments attached to the 6-methyluracyl moiety by the hexamethylene chains. Its silver complex possesses an abnormal high stability (logβ = 4.60 ± 0.16). Based on extraction and NMR data, we can conclude that the nitrogen donor atoms of 2-thiocytosine fragments in the investigated compounds participate in the coordination of Ag+ ion.
Experimental section
General remarks
All reagents and solvents were obtained from commercial sources. CHCl3 was distilled over P2O5. DMSO-d6 (99.5% isotopic purity), CDCl3 (99.8% isotopic purity) and CD3OD (99.8% isotopic purity) for NMR spectroscopy was used from Aldrich. The metal salts for extraction experiments were the following chlorides and nitrates: LiCl, NaCl, KCl, CsCl, CaCl2, CoCl2·6H2O, NiCl2, CuCl2, ZnCl2, AgNO3, CdCl2·2.5H2O, Hg(NO3)2·H2O, Pb(NO3)2, LaCl3·7H2O, Gd(NO3)3·6H2O and LuCl3·6H2O. Column chromatography was generally performed on Acros silica gel (pore size 60 Å, 40–63 μm particle size) and reactions were monitored by thin layer chromatography (TLC). Analysis was performed with Silufol-254 plates and visualized using UV light at 254 nm.Microanalyses of C, H and N were carried out with a CHN-3 analyzer. Melting points of compounds were measured with a Boetius hotstage apparatus. The MALDI-TOF mass spectra were measured on a Bruker ULTRAFLEX mass spectrometer (1,8,9-trihydroxyanthracene or 4-nitroaniline as the matrix). High resolution mass spectrometry data of the products were collected on a Finnigan MAT-212 instrument. The extraction process was studied by spectrophotometry on a Lambda 35 UV/VIS Spectrometer (Perkin Elmer Instruments) in quartz 0.5 cm cells with optical background correction. The IR absorption spectra were recorded in KBr pellets on a Vector-22 FT-IR spectrometer (Bruker) with a resolution of 4 cm−1 and 16 scan accumulation. The NMR spectra were detected on a Bruker spectrometers AVANCE-400 (400.1 MHz (1H), 100.6 MHz (13C)) and Bruker AVANCE-600 spectrometer equipped with a gradient block (field gradient to 50 G cm−1); working frequency of the spectrometer being 600.13 MHz in 1H, 150.90 MHz in 13C and 60.81 MHz in 15N experiments. A 5 mm inverse sensor with the gradient coil along the Z axis was used. The spectra were recorded at 303 ± 0.1 K. The chemical shifts are presented relatively to Me4Si as the internal standard for samples in CDCl3 and residual signals of DMSO for samples in CDCl3–DMSO-d6 (δ(DMSO-d6) = 2.50) or in CDCl3–CD3OD (δ(CD3OD) = 3.49).
Synthesis
Synthesis of pyrimidinophanes 7b–d, their acyclic counterparts 3, 4a,b and salts 9a,b was reported previously.23 1,3-Bis(6-bromohexyl)-6-methyluracil 10 (ref. 23) and model compounds 1 (ref. 55) and 2 (ref. 56 and 57) were synthesized by known procedures.:1 at −40 ÷ −45 °C a solution of meta-chloroperoxybenzoic acid (1.13 g, 4.6 mmol, 70% purity) in 50 ml of CHCl3–CH3OH 1:1 mixture was added dropwise during 1 h. The reaction mixture was stirred for 3 h and then washed with 150 ml of 10% NaHCO3 aqueous solution. The CHCl3 layer was separated, concentrated and subjected to column chromatography on SiO2. The column was eluted in succession with EtOAc and EtOAc–CH3OH 1:1 mixture. Elution with the solvent mixture gave 0.10 g (9%) of the disulfone 1,3-bis{4-[4-dimethylamino-6-methylpyrimidin-2-ylsulfonyl]butyl}-6-methyluracil (6) as an oil. 1H NMR (600.13 MHz, CDCl3–DMSO-d6 = 1:1 (v/v)) δ 1.70–1.90 (m, 8H, 4CH2), 2.22 (s, 3H, C(6)urCH3), 2.28 (s, 6H, 2C(6)pyrCH3), 3.08 (br. s, 12H, 4CH3), 3.41 (m, 4H, 2S(O)2CH2), 3.86 (m, 4H, 2NurCH2), 5.55 (s, 1H, C(5)urH), 6.01 (br. s, 2H, 2C(5)pyrH); 13C NMR (150.9 MHz, CDCl3–DMSO-d6 = 1:1 (v/v)) δ 18.9, 23.3, 27.8, 29.6, 36.3, 40.1, 44.2, 96.4, 100.4, 151.2, 161.1, 161.5, 164.0, 168.7; IR (neat, cm−1) ν = 2968, 2928, 2871, 1697, 1655, 1604, 1455, 1432, 1406, 1374, 1309, 1202, 1127, 1035, 970, 834, 770, 619. MALDI-MS (m/z): calcd for C27H40N8O6S2 [M + Na]+ 659.2, found: 559.3. Calcd for C27H40N8O6S2: C, 50.93; H, 6.33; N, 17.60; S, 10.07. Found: C, 50.91; H, 6.30; N, 17.62; S, 10.15%. Subsequent fractions of the solvent mixture afforded 0.75 g (71%) of the disulfoxide 5. Amorphous substance (decomp. > 77 °C); 1H NMR (600.1 MHz, CDCl3–DMSO-d6 = 1:1 (v/v)) δ 1.65–1.85 (m, 6H, 3CH2), 1.90–2.00 (m, 2H, CH2), 2.22 (s, 3H, C(6)urCH3), 2.43 (s, 6H, 2C(6)pyrCH3), 3.14 (br. s, 12H, 4CH3), 3.48 (m, 2S(O)CH2), 3.80 (m, 2H, 2N(1)urCH2), 3.90 (m, 2H, 2N(3)urCH2), 5.54 (s, 1H, C(5)urH), 6.24, 6.25 (both s, 1H each, 2C(5)pyrH); 13C NMR (150.9 MHz, CDCl3) δ 15.4, 17.6, 19.2, 19.8, 19.9, 20.0, 24.3, 26.7, 26.9, 28.1, 29.8, 37.4, 40.5, 40.7, 44.9, 50.6, 52.6, 53.2, 101.4, 101.5, 110.0, 151.2, 152.1, 162.2, 162.8, 166.3, 166.4, 171.0, 171.3; IR (KBr pellet, cm−1) ν = 2966, 2928, 2869, 1698, 1657, 1600, 1500, 1431, 1406, 1371, 1308, 1201, 1128, 1067, 1033, 968, 824, 769, 625. MALDI-MS (m/z): calcd for C27H40N8O4S2 [M + Na]+, [M + K]+ 627.5, 643.2, found: 627.2, 643.2. Calcd for C27H40N8O4S2: C, 53.62; H, 6.67; N, 18.53; S, 10.60. Found: C, 53.68; H, 6.70; N, 18.49; S, 10.65%.Synthesis of Ag+ complexes with ligands 1, 3, 4b and 7a
Our), 1661 ν(C(4)Ourl), 1603, 1500 ν(pyrimidine ring); MS MALDI-TOF, m/z: 629 [L4b + H]+, 651 [L4b + Na]+, 667 [L4b + K]+, 735 [Ag·L4b]+.Our), 1659 ν(C(4)Our), 1602, 1507 ν(pyrimidine ring); MS MALDI-TOF, m/z: 613 [L7a + H]+, 719 [Ag·L7a]+.X-ray crystallographic study
Crystallographic data for derivative 7c (C34H52N8O2S2): M = 668.96, monoclinic system, space group P21, a = 7.347 (1) Å, b = 19.924 (4) Å, c = 13.152 (2) Å, β = 100.454 (2)°, Z = 4, V = 1893.1(6) Å3, ρcalc = 1.174 g cm−3, μ(Mo-Kα) = 0.71073 Å, crystal dimensions of 0.38 × 0.24 × 0.11 mm. Data were collected at 150(2) K on Smart Apex II automatic diffractometer using graphite monochromated radiation. The structures were solved by direct methods and refined by full-matrix least-squares using the SHELXL97 program58 to final R = 0.0705 and Rw = 0.2279 using 7272 independent reflections (Θmax = 26.00°) and 403 parameters. All the non-hydrogen atoms were refined with anisotropic atomic displacement parameters. The hydrogen atoms were placed in calculated positions. All figures were made using the program Mercury.†59 The structure was deposited into the Cambridge Structural Database under number CCDC 973067.Biological assay
The in vitro antibacterial and antifungal activity of the synthesized compounds were investigated against Pseudomonas aeruginosa 9027, Escherichia coli F-50, Staphylococcus aureus 209p, Bacillus subtilis 6633 and yeast Candida albicans 885-653. Minimal inhibitory concentrations were estimated by conventional dilution methods for bacteria60 and fungi.61Acknowledgements
This work is supported by the Russian Fund for Basic Research (grants 13-03-00709, 13-04-40288-H and 13-03-97073 Tatarstan).Notes and references
- M. Radi, S. Schenone and M. Botta, Org. Biomol. Chem., 2009, 7, 2841–2847 CAS.
- M. D. Hall and T. V. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- E. Freisinger and R. K. O. Sigel, Coord. Chem. Rev., 2007, 251, 1834–1851 CrossRef CAS PubMed.
- Interplay between metal ions and nucleic acid, ed. A. Sigel, H. Sigel and R. K. O. Sigel, Springer, Dordrecht, Heidelberg, London, New York, 2012, vol. 10 of Metal ions in life sciences Search PubMed.
- T. Lönnberg and M. Luomala, Org. Biomol. Chem., 2012, 10, 6785–6791 Search PubMed.
- A. M. Pyle, Science, 1993, 261, 709–714 CAS.
- The RNA world, ed. R. F. Gesteland, T. R. Cech and J. F. Atkins, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 2nd edn, 1999, vol. 37, pp. 287–319 Search PubMed.
- V. J. DeRose, S. Burns, N. K. Kim and M. Vogt, Compr. Coord. Chem. II, 2004, 8, 787–813 CAS.
- A. Ono, H. Togiroe, Y. Tanaka and I. Okamoto, Chem. Soc. Rev., 2011, 40, 5855–5866 RSC.
- J. M. Lehn, Supramolecular chemistry. Concepts and perspectives, VCH, Weinheim, 1995 Search PubMed.
- Transition metals in supramolecular chemistry, ed. J. P. Sauvage, Wiley, Chichester, 1999 Search PubMed.
- G. R. Desiraju and T. Steiner, The weak hydrogen bond in structural chemistry and biology, Oxford University Press, Oxford, 1999 Search PubMed.
- M.-Y. Pan, W. Hang, X.-J. Zhao, H. Zhao, P.-C. Deng, Z.-H. Xing, Y. Qing and Y. He, Org. Biomol. Chem., 2011, 9, 5692–5702 CAS.
- G. Beobide, O. Castillo, J. Cepeda, A. Luque, S. Pérez-Yáñez, P. Román and J. Thomas-Gipson, Coord. Chem. Rev., 2013, 257, 2716–2736 CrossRef CAS PubMed.
- B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487–516 CrossRef CAS.
- J. A. R. Navarro and B. Lippert, Coord. Chem. Rev., 2001, 222, 219–250 CrossRef CAS.
- J. J. Fiol, M. Barceló-Oliver, A. Tasada, A. Frontera, À. Terrón and À. García-Raso, Coord. Chem. Rev., 2013, 257, 2705–2715 CrossRef CAS PubMed.
- M. Goodgame and D. A. Jakwbovic, Coord. Chem. Rev., 1987, 79, 97–134 CrossRef CAS.
- V. E. Semenov, J. Inclusion Phenom. Macrocyclic Chem., 2013, 77, 1–22 CrossRef CAS.
- M. M. Htay and O. Meth-Cohn, Tetrahedron Lett., 1976, 17, 469–472 CrossRef.
- J. T. Redd, J. S. Bradshaw, P. Huszthy and R. M. Izatt, J. Inclusion Phenom. Macrocyclic Chem., 1997, 29, 301–308 CrossRef CAS.
- J. S. Bradshaw, P. Huszthy, J. Ty Redd, X.-X. Zhang, T.-M. Wang, J. K. Hathaway, J. Young and R. M. Izatt, Pure Appl. Chem., 1995, 67, 691–695 CrossRef CAS.
- V. E. Semenov, A. S. Mikhailov, A. D. Voloshina, N. V. Kulik, A. D. Nikitashina, V. V. Zobov, S. V. Kharlamov, S. K. Latypov and V. S. Reznik, Eur. J. Med. Chem., 2011, 46, 4715–4724 CrossRef CAS PubMed.
- A. S. Mikhailov, R. Kh. Giniyatullin, V. E. Semenov, V. S. Reznik, A. A. Nafikova, Sh. K. Latypov, Y. Y. Efremov and D. R. Sharafutdunova, Russ. Chem. Bull., 2003, 52, 1399–1402 CrossRef CAS.
- R. R. Shagidullin, A. V. Chernova, G. M. Doroshkina, V. E. Kataev, Z. G. Bazhanova, S. A. Katsyuba, V. S. Reznik, A. S. Mikhailov, R. Kh. Giniyatullin, N. G. Pashkurov, Yu. Ya. Efremov and A. A. Nafikova, Russ. J. Gen. Chem., 2002, 72, 1625–1632 CrossRef CAS.
- V. E. Semenov, V. I. Morozov, A. V. Chernova, R. R. Shagidullin, A. S. Mikhailov, R. Kh. Giniyatullin, V. D. Akamsin and V. S. Reznik, Russ. J. Coord. Chem., 2007, 33, 685–691 CrossRef CAS.
- J. Carbon, H. David and M. H. Studier, Science, 1968, 161, 1146–1147 CAS.
- R. A. Roosaf and E. D. DeLamater, Cancer Res., 1960, 20, 1543–1554 Search PubMed.
- H. Rostkowska, M. J. Novak, L. Lapiński, M. Bretner, T. Kulikowski, A. Les and L. Adamowicz, Biochim. Biophys. Acta, 1993, 1172, 239–246 CrossRef CAS.
- J. Timson, D. J. Price and J. S. Walker, Cytobios, 1972, 5, 97–100 CAS.
- K. Singh, B. Groth-Vasselli, P. N. Farnsworth and D. K. Rai, Res. Commun. Mol. Pathol. Pharmacol., 1996, 94, 129–140 CAS.
- T. Kawaguchi, T. Ichikawa, T. Hasegawa, M. Saneyoshi, A. Yukita, M. Asano, T. Wakayama, H. Kato and T. Nagata, Biol. Pharm. Bull., 1999, 22, 100–102 CAS.
- T. Kawaguchi, T. Ichikawa, T. Hasegawa, M. Saneyoshi, T. Wakayama, H. Kato, A. Yukita and T. Nagata, Chem. Pharm. Bull., 2000, 48, 454–457 CrossRef CAS.
- C. Vetter, Ch. Wagner, G. N. Kaluđerović, B. R. Paschke and D. Steinborn, Inorg. Chim. Acta, 2009, 362, 189–195 CrossRef CAS PubMed.
- S. Shigeta, S. Mori, T. Kira, K. Takahashi, E. Kodama, K. Konno, T. Nagata, H. Kato, T. Wakayama, N. Koike and M. Saneyoshi, Antiviral Chem. Chemother., 1999, 10, 195–209 CAS.
- D. P. Bhave, W. B. Muse and K. S. Carroll, Infect. Disord.: Drug Targets, 2007, 7, 140–158 CrossRef CAS.
- S. N. Podyachev, S. N. Sudakova, V. V. Syakaev, A. K. Galiev, R. R. Shagidullin and A. I. Konovalov, Supramol. Chem., 2008, 20, 479–486 CrossRef CAS.
- S. N. Podyachev, N. E. Burmakina, V. V. Syakaev, S. N. Sudakova, R. R. Shagidullin and A. I. Konovalov, Tetrahedron, 2009, 65, 408–417 CrossRef CAS PubMed.
- R. J. W. Lugtenberg, M. M. G. Antonisse, R. J. M. Egberink, J. F. J. Engbersen and D. N. Reinhoudt, J. Chem. Soc., Perkin Trans. 2, 1996, 1937–1941 RSC.
- J. Xie, Q.-Y. Zheng, Y.-S. Zheng, C.-F. Chen and Z.-T. Huang, J. Inclusion Phenom. Macrocyclic Chem., 2001, 40, 125–130 CrossRef CAS.
- A. T. Yordanov, D. M. Roundhill and J. T. Mague, Inorg. Chim. Acta, 1996, 250, 295–302 CrossRef CAS.
- S. Bouhroum, J. S. Kim, S. W. Lee, P. Thuéry, G. Yap, F. Arnaud-Neu and J. Vicens, J. Inclusion Phenom. Macrocyclic Chem., 2008, 62, 239–250 CrossRef CAS.
- A. T. Yordanov, O. M. Falana, H. F. Koch and D. M. Roundhill, Inorg. Chem., 1997, 36, 6468–6471 CrossRef CAS.
- T. M. Buslaeva, E. A. Krylova, E. V. Volchkova, S. P. Gromov and N. I. Sidorenko, Russian Journal of Non-Ferrous Metals, 2008, 49, 459–465 CrossRef.
- T. M. Buslaeva, E. A. Krylova, E. V. Volchkova, S. P. Gromov and N. I. Sidorenko, Russian Journal of Non-Ferrous Metals, 2009, 50, 461–470 CrossRef.
- T. M. Buslaeva, E. A. Krylova, E. V. Volchkova, S. P. Gromov and N. I. Sidorenko, Russian Journal of Non-Ferrous Metals, 2010, 51, 457–466 CrossRef.
- D. M. Roundhill, I. B. Solangi, S. Memon, M. I. Bhanger and M. Yilmaz, Pak. J. Anal. Environ. Chem., 2009, 10, 1–13 CAS.
- V. Arora, H. M. Chawla and S. P. Singh, ARKIVOC, 2007, ii, 172–200 CrossRef.
- A. T. Yordanov, B. R. Whittlesey and D. M. Roundhill, Inorg. Chem., 1998, 37, 3526–3531 CrossRef CAS PubMed.
- S. N. Podyachev, N. E. Kashapova, V. V. Syakaev, S. N. Sudakova, R. R. Zainullina, M. Gruner, W. D. Habicher, T. A. Barsukova, F. Yang and A. I. Konovalov, J. Inclusion Phenom. Macrocyclic Chem., 2014, 78, 371–380 CrossRef CAS PubMed.
- R. R. Shagidullin, A. V. Chernova, Z. G. Bazhanova, J.-H. Lii, V. E. Kataev, S. A. Katsyuba and V. S. Reznik, J. Mol. Struct., 2004, 707, 1–9 CrossRef CAS PubMed.
- I. Okamoto, T. Ono, R. Sameshima and A. Ono, Chem. Commun., 2012, 48, 4347–4349 RSC.
- C. N. Banti and S. K. Hadjikakou, Metallomics, 2013, 5, 569–596 RSC.
- W. K. Jung, H. C. Koo, K. W. Kim, S. Shin, S. H. Kim and Y. H. Park, Appl. Environ. Microbiol., 2008, 74, 2171–2178 CrossRef CAS PubMed.
- E. A. Arutyunyan, V. I. Gunar, E. P. Gracheva and S. I. Zav'yalov, Izv. Akad. Nauk SSSR, Ser. Khim., 1969, 18, 587–592 Search PubMed.
- D. S. Snyder, L. Tradtrantip, C. Yao, M. J. Kurth and A. S. Verkman, J. Med. Chem., 2011, 54, 5468–5477 CrossRef CAS PubMed.
- M. Prystas and F. Sorm, Collect. Czech. Chem. Commun., 1969, 34, 2316–2347 CrossRef CAS.
- G. M. Sheldrick, SHELX-97. Programs for Crystal Structure Analysis (Release 97-2), University of Göttingen, Germany, 1997 Search PubMed.
- I. J. Bruno, J. C. Cole, P. R. Edgington, M. Kessler, C. F. Macrae, P. McCabe, J. Pearson and R. Taylor, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 389–397 CrossRef PubMed.
- National Committee for Clinical Laboratory Standards, Methods for dilution antimicrobial susceptibility. Tests for bacteria that grow aerobically, Wayne, PA, USA, 6th edn: Approved standard, M7–A5, NCCLS, 2000 Search PubMed.
- National Committee for Clinical Laboratory Standards, Reference method for broth dilution antifungal susceptibility testing of conidium-forming filamentous fungi, Wayne, PA, USA, Proposed standard, M38-P, NCCLS, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Extraction conditions, 1H and 15N NMR data and minimal inhibitory concentrations. CCDC 973067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47571a |
| This journal is © The Royal Society of Chemistry 2014 |