
A new approach to rubber reinforcement
Abstract
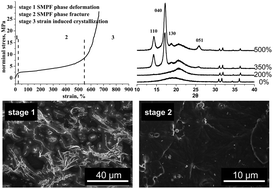
In this paper, we report the highly efficient reinforcement of dynamically cross-linked silane-modified phenol formaldehyde resin (SMPF) in ethylene-propylene diene terpolymer rubber (EPDM). Only 5 phr of SMPF is able to greatly improve the tensile strength of EPDM from 5 MPa to 28 MPa, which is 55% higher than that of EPDM filled with 30 phr of carbon black N330, and it still retains a high elongation of more than 600%. The scanning electron microscopy and X-ray diffraction results reveal the reinforcement mechanism of EPDM. SMPF is uniformly dispersed in the EPDM matrix by a conventional Banbury mixer and is dynamically cross-linked to form a greatly expanded hard phase comprising the interpenetrating polymer networks of EPDM and SMPF during the mixing process, which leads to a high modulus. The high tensile strength is attributed to the strain-induced crystallization of EPDM at high strain according to the X-ray diffraction patterns of the stretched EPDM–SMPF composites. These results reveal the high efficient reinforcement of the thermoset resin in rubber, demonstrate a new mechanism of rubber reinforcement and suggest a new direction for rubber reinforcement.
 Please wait while we load your content...
Please wait while we load your content...

