
Nanofibrous electroactive scaffolds from a chitosan-grafted-aniline tetramer by electrospinning for tissue engineering
Abstract
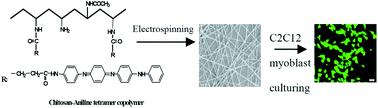
Functional degradable biomimetic scaffolds have great potential applications in tissue regeneration. Nanofibrous electroactive biodegradable scaffolds from chitosan-grafted-aniline tetramer (CS–AT) were fabricated by an electrospinning method. The CS–AT was synthesized by amidation reaction between the carboxyl group of aniline tetramer and the amine group of chitosan. The structure of CS–AT copolymer was characterized by 1H NMR, FT-IR, TGA and XRD. UV-vis and cyclic voltammetry tests were used to demonstrate the electroactivity of CS–AT. The electrospun nanofibers were created from CS–AT solution. The morphology of the CS–AT nanofibers was observed by employing SEM and the results illustrated that the diameter of the nanofibers of deposited CS and CS–AT samples was controlled by both polymer concentration and the AT content. The biocompatibility of the materials was evaluated by cell adhesion and proliferation of C2C12 myoblasts and dog chondrocyte cells, and the results demonstrated that the CS–AT materials had good biocompatibility and greatly enhanced the cell adhesion and proliferation of C2C12 cells.
- This article is part of the themed collection: Chemistry for Medicine: Special Collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

