 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Visualisation and characterisation of heterogeneous bimodal PDMS networks†
F. B.
Madsen
,
A. E.
Daugaard
,
C.
Fleury
,
S.
Hvilsted
and
A. L.
Skov
*
Danish Polymer Centre, Department of Chemical and Biochemical Engineering, Technical University of Denmark, DTU, Søltofts Plads, Bldg. 227, 2800 Kgs. Lyngby, Denmark. E-mail: al@kt.dtu.dk
First published on 6th January 2014
Abstract
The existence of short-chain domains in heterogeneous bimodal PDMS networks has been confirmed visually, for the first time, through confocal fluorescence microscopy. The networks were prepared using a controlled reaction scheme where short PDMS chains were reacted below the gelation point into hyperbranched structures using a fluorescent silicone compatible cross-linker. The formation of the hyperbranched structures was confirmed by FTIR, 1H-NMR and size exclusion chromatography (SEC). The short-chain hyperbranched structures were thereafter mixed with long-chain hyperbranched structures to form bimodal networks with short-chain domains within a long-chain network. The average sizes of the short-chain domains were found to vary from 2.1 to 5.7 μm depending on the short-chain content. The visualised network structure could be correlated thereafter to the elastic properties, which were determined by rheology. All heterogeneous bimodal networks displayed significantly lower moduli than mono-modal PDMS elastomers prepared from the long polymer chains. Low-loss moduli as well as low-sol fractions indicate that low-elastic moduli can be obtained without compromising the network's structure.
Introduction
Polydimethylsiloxane (PDMS) elastomers are known for their unique properties, such as high extensibility, high thermal stability, low surface tension and chemical and biochemical inertness.1 PDMS elastomers consist of a PDMS network, usually resulting from hydrosilylation, radical or condensation reactions, silica particles and other fillers.2 PDMS networks without fillers suffer from low tear strength and are therefore not easily handled as thin films. The reinforcing silica particles in the elastomer formulations result in a very weak but still not negligible optical distortion, which constitutes a problem in many commercial applications such as adaptable lenses and optical biochips.3,4The mechanical properties of PDMS elastomers, as an alternative, can be improved by creating bimodal networks which are prepared by applying advanced mixing schemes. Bimodal networks consist of very short and relatively long chemically identical polymer chains cross-linked into a network, and their mechanical properties are superior to traditional elastomers.5,6 Several studies have investigated the unexpected high tear resistance and ultimate strength of bimodal networks.7–9 The elastic modulus of bimodal networks increases significantly at high elongations, so the ultimate strength of the networks is very high. Improvements in ultimate strength stem from the limited extensibility of the short chains, which gives the networks toughness.10,11 Furthermore, the relatively long chains retard the rupture process and provide extensibility to the network.9 Therefore, bimodal networks exhibit both a substantial ultimate stress and ultimate strain.6 Bimodal networks can be created by the random distribution of short polymer chains within the long chains,6,12–14 or as heavily cross-linked short-chain domains joined to the long-chain network.15–17 Bimodal networks with homogenous (random) distributions of short and long chains are prepared in one-step procedures where the chains are mixed and cross-linked simultaneously to form random networks. In contrast, heterogeneous bimodal networks are prepared in two-step procedures. The short-chain domains are prepared in a reaction that happens prior to the cross-linking, where the short chains are reacted below the gelation threshold into hyperbranched structures which are subsequently mixed and cross-linked together with the long chains. Consequently, short-chain domains are created within the long-chain network. Furthermore, the long chains can also be reacted with cross-linker molecules below the gelation threshold – prior to the network formation – in order to obtain even higher control over the network and the distribution of long- and short-chain domains.17
We have previously demonstrated that the advantage of forming heterogeneous bimodal networks with a controlled distribution of short-chains is that the elastic modulus becomes lower compared to both homogenous bimodal networks with similar short chain concentrations and mono-modal PDMS networks.17 In so doing, softer and more stretchable PDMS elastomers are obtained. The films are at the same time easily handled as thin films, which for the homologues mono-modal PDMS network is difficult without the addition of silica particles. Furthermore, heterogeneous bimodal networks have been shown to exhibit very low viscous losses in combination with low elastic moduli.17 So they are therefore excellent alternatives to both mono-modal PDMS elastomers, which require fillers to have sufficiently high tear strength to make them manageable as thin films, and to homogenous bimodal networks, which have higher elastic moduli due to the random distribution of short chains.
We herein present a novel method for analysing the size of short-chain domains in heterogeneous bimodal PDMS networks. A recently developed silicone compatible cross-linker allowing for orthogonal chemistry18 is used to tag the short-chain hyperbranched structures at their cross-linking points with fluorescent molecules. The size of the short-chain domains will furthermore be correlated to the final mechanical properties of the networks. The design scheme offers an unprecedented way of tuning the properties of heterogeneous bimodal networks and thereby paves the way for the implementation of such networks in advanced applications where strong, unfilled and largely extensible networks are needed. The properties of the resulting networks will fulfil the demands of many commercial PDMS elastomer applications, including soft lithography applications where higher fracture energy is needed,19 optical applications where strong elastomers without optically distorting fillers are desired3,4 and dielectric elastomers where soft networks with high extensibilities and low viscous losses are essential.20–25
Experimental
General methods
FTIR analyses were conducted on a Perkin-Elmer Spectrum One model 2000 Fourier Transform Infrared apparatus equipped with a universal attenuated total reflection accessory on a ZnSe–diamond composite. The spectra were recorded in the range of 4000–650 cm−1 with 16 scans using 4 cm−1 resolution. 1H-NMR spectra were recorded on a Bruker 300 MHz spectrometer. Size-exclusion chromatography (SEC) was performed on a Viscotek GPCmax VE-2001 instrument, equipped with a Viscotek TriSEC Model 302 triple detector using two PLgel mixed-D columns, provided by Polymer Laboratories. Samples were run in THF at a rate of 1 ml min−1, and molar mass characteristics were calculated using polystyrene standards. Optical characterisations were performed on a Leica TCS SPE high-resolution confocal microscope with a 10× lens at 400 nm in the z-direction, equipped with a Leica CTR4000 electronic box. Image analysis was performed with ImageJ, and the domain diameter was taken as an average over 100 domains. Differential scanning calorimetry (DSC) measurements were performed on a DSC Q1000, provided by TA Instruments. The thermal analyses were performed with a heating and cooling rate of 10 °C min−1, from −90 to 150 °C. Thermogravimetric analysis (TGA) was performed on a Q500 (TA Instruments) in a nitrogen atmosphere with a heating rate of 10 °C min−1, from RT to 950 °C. All degradation temperatures were taken as the peak temperature of the first derivative of the temperature with time. Linear rheology of the bimodal films was performed with a TA Instruments TA 2000 Rheometer set to a controlled strain mode of 1%, which was ensured to be within the linear viscoelastic regime. The measurements were performed in the frequency range 100–0.01 Hz with a parallel plate geometry of 25 mm. The films were approximately 1 mm thick and were measured with a small normal force from the upper plate, which ensured that there were no-slip conditions.Materials
Hydride-terminated PDMS: DMS-H11 (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n = 1050 g mol−1) and DMS-H31 (n = 28
n = 1050 g mol−1) and DMS-H31 (n = 28![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) as well as a vinyl functional PDMS cross-linker, VDT-431 (n = 28000 g mol−1, ∼16 vinyl groups), were acquired from Gelest Inc. The platinum cyclovinylmethyl siloxane complex catalyst (511) was provided by Hanse Chemie. All other chemicals were acquired from Sigma-Aldrich and used as received, unless otherwise specified.
000 g mol−1) as well as a vinyl functional PDMS cross-linker, VDT-431 (n = 28000 g mol−1, ∼16 vinyl groups), were acquired from Gelest Inc. The platinum cyclovinylmethyl siloxane complex catalyst (511) was provided by Hanse Chemie. All other chemicals were acquired from Sigma-Aldrich and used as received, unless otherwise specified.
Fluorescent cross-linker: (4-methyl-7-((1-(3-(tris(allyloxy)silyl)propyl)-1H-1,2,3-triazol-4-yl)methoxy)-2H-chromen-2-one)
The fluorescent tri-functional cross-linker (3) was prepared according to previously described procedures.18 A silicone compatible tri-functional azide cross-linker (1) was synthesised via the silyl ether reaction between (3-bromopropyl)trichlorosilane and allyl alcohol and the subsequent substitution of bromine with azide. The azide-functional cross-linker was thereafter used in a click reaction (the copper-catalysed cyclo-addition of an azide and an alkyne (CuAAC) forming a 1,4-disubstituted-1,2,3-triazole) with the alkyne 4-methyl-7-(prop-2-yn-1-yloxy)-2H-chromen-2-one (2), which was prepared through a Williamson ether synthesis of 4-methylumbelliferone and propargyl bromide.

General procedure for preparing heterogeneous bimodal networks (amounts given for 10 wt% short chains)
The fluorescent tri-functional cross-linker (3, 0.027 g, 0.0054 mmol) was dissolved in THF (0.6 ml) and mixed with short PDMS chains (DMS-H11, 1050 g mol−1, 0.212 g, 0.020 mmol). The platinum cyclovinylmethyl siloxane complex catalyst (511) (10 ppm) in dry THF was thereafter added to give a stoichiometry of r = 0.4 and the mixture was mixed in a FlackTek Inc. DAC 150.1 FVZ-K SpeedMixer™ and allowed to react for 1 hour at 60 °C. Next, a 16-functional vinyl cross-linker (VDT-431, 28000 g mol−1, 0.014 g, 5 × 10−5 mmol) and the long PDMS chains (DMS-H31, 28000 g mol−1, 1.91 g, 0.0068 mmol) were mixed with platinum cyclovinylmethyl siloxane complex catalyst (511) (10 ppm) in dry THF to give a stoichiometry of r = 0.06. The mixture was combined in the SpeedMixer and thereafter allowed to react for 1 hour at 60 °C. After the initial reactions, the short- and long-chain mixtures were combined and additional cross-linker (VDT-431, 28000 g mol−1, 0.838 g, 0.0030 mmol) and catalyst (10 ppm) were added to give an overall stoichiometry of r = 1.2 (20% excess of vinyl groups). The mixture was blended in the SpeedMixer and poured into 1 mm thick steel moulds on an FEP fluorocarbon substrate, degassed in vacuo for 1 hour, cured for 24 hours at RT and then post-cured at 60 °C for 1 hour. Short and long chain amounts were varied to create bimodal networks with mass ratios of short:long chains = 0:100, 3:97, 10:90, 20:80 and 30:70, corresponding to molar ratios of short:long chains = 0:100, 45:55, 75:25, 87:13 and 92:8.
Swelling experiments
Swelling experiments were performed by placing a pre-weighed sample in a flask containing toluene until the equilibrium swelling state was reached (typically one week). At the end of the swelling period, the samples were removed from the solvent, dried quickly and carefully with lint-free tissue paper and then weighed. The volume degree of equilibrium swelling (Qv) was then calculated according to: Qv = 1 + (Qw − 1) × νtoluene/νPDMS, where νtoluene is the specific volume of toluene and νPDMS is the specific volume of PDMS. Qw is calculated as: Qw = m0/m, where m0 is the initial weight of the (dry) sample before swelling and m is the weight of the wet (swollen) sample. After the swelling experiment, the samples were washed several times with toluene and dried for 24 hours. The sol fractions (Wsol) were thereafter calculated as: Wsol(%) = (1 − (me/m0)) × 100, where me is the weight of the dry sample after the swelling/extraction.Results and discussion
The heterogeneous bimodal PDMS networks were prepared in a two-step procedure combining short- and long-chain hyperbranched PDMS structures, as illustrated in Fig. 1. First, fluorescent short-chain hyperbranched structures were prepared in a reaction prior to the network formation using a previously prepared fluorescent tri-functional cross-linker.18 The cross-linker was used in concentrations below the gelation point, as determined by the Flory–Stockmayer expression: rc = 1/(f − 1) where rc is the stoichiometry between the reactive groups on the cross-linker and the reactive groups on the polymer required for critical gelation, and f is the functionality of the cross-linker. By using a stoichiometry where r < rc no infinite short-chain network will be created, and the formed hyperbranched structures still have reactive end groups.26,27 The short-chain hyperbranched structures were characterised by FTIR, 1H-NMR and size-exclusion chromatography (SEC). The reaction between the fluorescent cross-linker and the short PDMS chains was followed by FTIR and the decreasing intensity of the cross-linker C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C band at 1610 cm−1. Furthermore, FTIR spectra confirmed the presence of the remaining Si–H end groups (at 2125 cm−1) on the short-chain polymers after the reaction. The reaction employed to form the short-chain hyperbranched structures was also followed with 1H-NMR by the disappearance of the –CH2CH2 resonance at 5.26 ppm and –CH2CH resonance at 5.12 ppm. The presence of Si–H protons at 4.70 ppm after the reaction indicates that reactive polymer end groups are indeed still present on the hyperbranched structures. From the areas under the Si–H resonance before and after the reaction relative to the areas of the constant Si–CH3 resonances at 0.06–0.18 ppm it can be deduced that ∼60% reactive Si–H end groups remain after the reaction. This number is consistent with the stoichiometric ratio of r = 0.4 used in this instance. 1H-NMR spectra can be found as ESI.† SEC was used to confirm the post-reaction increase in molecular weight of the hyperbranched structures, which appear in the higher molecular weight region compared to the low molecular weight short-chain PDMS (as seen in the SEC traces in the ESI†). In addition, the SEC trace clearly shows that the reaction results in a distribution of hyperbranched structures and not in a mono-modal structure, which is as expected for a hyperbranched structure.
C band at 1610 cm−1. Furthermore, FTIR spectra confirmed the presence of the remaining Si–H end groups (at 2125 cm−1) on the short-chain polymers after the reaction. The reaction employed to form the short-chain hyperbranched structures was also followed with 1H-NMR by the disappearance of the –CH2CH2 resonance at 5.26 ppm and –CH2CH resonance at 5.12 ppm. The presence of Si–H protons at 4.70 ppm after the reaction indicates that reactive polymer end groups are indeed still present on the hyperbranched structures. From the areas under the Si–H resonance before and after the reaction relative to the areas of the constant Si–CH3 resonances at 0.06–0.18 ppm it can be deduced that ∼60% reactive Si–H end groups remain after the reaction. This number is consistent with the stoichiometric ratio of r = 0.4 used in this instance. 1H-NMR spectra can be found as ESI.† SEC was used to confirm the post-reaction increase in molecular weight of the hyperbranched structures, which appear in the higher molecular weight region compared to the low molecular weight short-chain PDMS (as seen in the SEC traces in the ESI†). In addition, the SEC trace clearly shows that the reaction results in a distribution of hyperbranched structures and not in a mono-modal structure, which is as expected for a hyperbranched structure.
 | ||
| Fig. 1 Schematic illustration of the two-step reaction procedure employed to form heterogeneous bimodal networks. | ||
In a similar manner, the long-chain hyperbranched structures were prepared by using a commercial cross-linker (28000 g mol−1, bearing, on average, 16 vinyl groups per chain) and a higher molecular weight PDMS (28000 g mol−1). The hyperbranched structures, based on the short and long chains, were mixed and cross-linked by using the commercial cross-linker in excess. The fluorescent and reinforcing short-chain hyperbranched structures thereby became covalently bound to the long-chain hyperbranched structures, ensuring sufficient separation of the short-chain structures to maximise the reinforcing effect.
The heterogeneous bimodal networks were prepared with varying contents of short-chain hyperbranched structures, in order to illustrate the effect of the ratio between long- and short-chain hyperbranched structures. The concentration of short-chain structures varied from 0 to 30 wt%, corresponding to 0 to 92 mol% (of the total number of hydride functional polymers). Films based entirely on the short-chain polymer could not be prepared, as the polymer was of a too low molecular weight to form a mechanically stable film.
The prepared heterogeneous bimodal networks were characterised by confocal microscopy, which uses single-point illumination and rejects out-of-focus light. Thus, images with better vertical optical resolution than traditional fluorescence microscopy are obtained. The acquired images had reduced background haze and represent a thin cross-section of the sample, as shown in Fig. 2 (original fluorescence confocal microscopy images of all samples can be found as ESI†).
 | ||
| Fig. 2 Grayscale confocal microscopy images obtained at 400 nm of the prepared heterogeneous bimodal networks with varying concentrations of short-chain hyperbranched structures. (a) 3 wt% and (b) 30 wt%. | ||
The fluorescent domains are evenly distributed within all networks. Furthermore, it can be seen from Fig. 2 that the fluorescent domains vary in size and abundance, depending on the ratio between the short- and long-chain hyperbranched structures in the network. The mean diameters of the fluorescent domains were determined as an average of 100 domains, the results for which are summarised in Table 1. As seen in Table 1, the sizes of the fluorescent domains vary only moderately for the concentrations of short-chain hyperbranched structures, from 3 wt% to 20 wt%, whereas substantially larger domains are observed at 30 wt%. Furthermore, it is clear from Fig. 2 that the domains are very well distributed, and the number of domains is high for both samples. When the concentration of the short-chain hyperbranched structures is increased to 30 wt%, a substantial increase in the domain's size is evident (Fig. 2), due to the short-chain hyperbranched structures becoming so tightly packed that even upon ideal distribution they will interconnect covalently and turn into larger agglomerates. This is also substantiated by the large increase in the standard deviation of the measured average short-chain domain diameter.
| Short-chain concentration | Domain diametera (D) | C N b × 10−9 | G′@1 Hz | G′′@1 Hz | |
|---|---|---|---|---|---|
| [Wt%] | [Mol%] | [μm] | [g−1] | [kPa] | [kPa] |
| a The mean diameters were determined with the imaging processing program ImageJ. b The number average concentration of short-chain domains is calculated as: CN = ((6 × wt%)/(πD3))(1/ρPDMS), where ρPDMS is the density of PDMS. | |||||
| 0 | 0 | — | — | 93.4 | 1.19 |
| 3 | 45 | 2.9 ± 0.9 | 2.4 | 11.8 | 0.32 |
| 10 | 75 | 2.8 ± 0.8 | 9.0 | 21.5 | 0.20 |
| 20 | 87 | 2.1 ± 0.5 | 43 | 78.1 | 0.98 |
| 30 | 92 | 5.7 ± 1.6 | 3.2 | 59.5 | 0.70 |
The influence on the distribution and size of the observed domains on the elastic and mechanical properties of the bimodal networks was determined by employing small amplitude rheology. The resulting shear storage moduli (G′) and loss moduli (G′′) are summarised in Table 1. The mono-modal PDMS reference network, with 0% of short chains, has a storage modulus of 93.4 kPa. Upon the addition of the short-chain hyperbranched structures, the modulus decreases significantly to 11.8 kPa. From fundamental theories on rubber elasticity the elastic moduli are expected to scale linearly with the inverse average molecular weight of the polymer chains in the network.28 As such, the addition of very short polymer chains to the system would be expected to decrease the average molecular weight of the system and thus increase the elastic modulus significantly. This decrease in the elastic modulus in heterogeneous bimodal networks has been shown previously but not explained with supporting data.16,17 Based on the unprecedented correlation between the network composition, size and distribution of the fluorescent domains, and the elastic properties of heterogeneous bimodal networks, we believe that the decrease in the elastic modulus can be explained by a multi-domain theory. When short-chain hyperbranched structures are added, they naturally contain a large concentration of unreacted hydride groups on the surface. These groups react with cross-linker molecules, which upon initial reaction are hindered from diffusion by reptation.29 Consequently, there is a large concentration of cross-linker molecules on the surface of the hyperbranched structures. Small cross-linker molecules, such as those used in our previous study,17 are packed tightly around the surface, whereas the long-chain cross-linker molecules used in this study tend to fold around the short-chain hyperbranched structures. In both cases, this creates areas around the cross-linker molecules where the effective cross-linking density is lower than in the bulk long-chain network, as a large number of the cross-linking sites are present around the short-chain domains, as illustrated in Fig. 3. In this study, with a high molecular weight cross-linker, folding back to the short-chain domains also leads to the formation of loops, which do not contribute to elasticity to the same extent as the true cross-linking sites.30 Domains with surrounding cross-linker molecules therefore create a local softening effect in the PDMS networks. The heterogeneous bimodal elastomers will consequently be softer and have a lower elastic modulus than the homologous mono-modal networks despite higher average cross-linking density. The reduction in the modulus, from mono-modal to a small amount of short-chain domains, is confirmed experimentally. The elastic modulus is then increased with increasing short-chain concentration. At high concentrations of short chains the elastic modulus drops again. This can be explained by a drop in the number average concentration of short-chain domains CN = ((6 × wt%)/(πD3))(1/ρPDMS), with the additional increase of short chains (see Table 1). From the calculations it can be seen that for the bimodal networks, the elastic modulus increases with increasing CN, although, not in a trivial way.
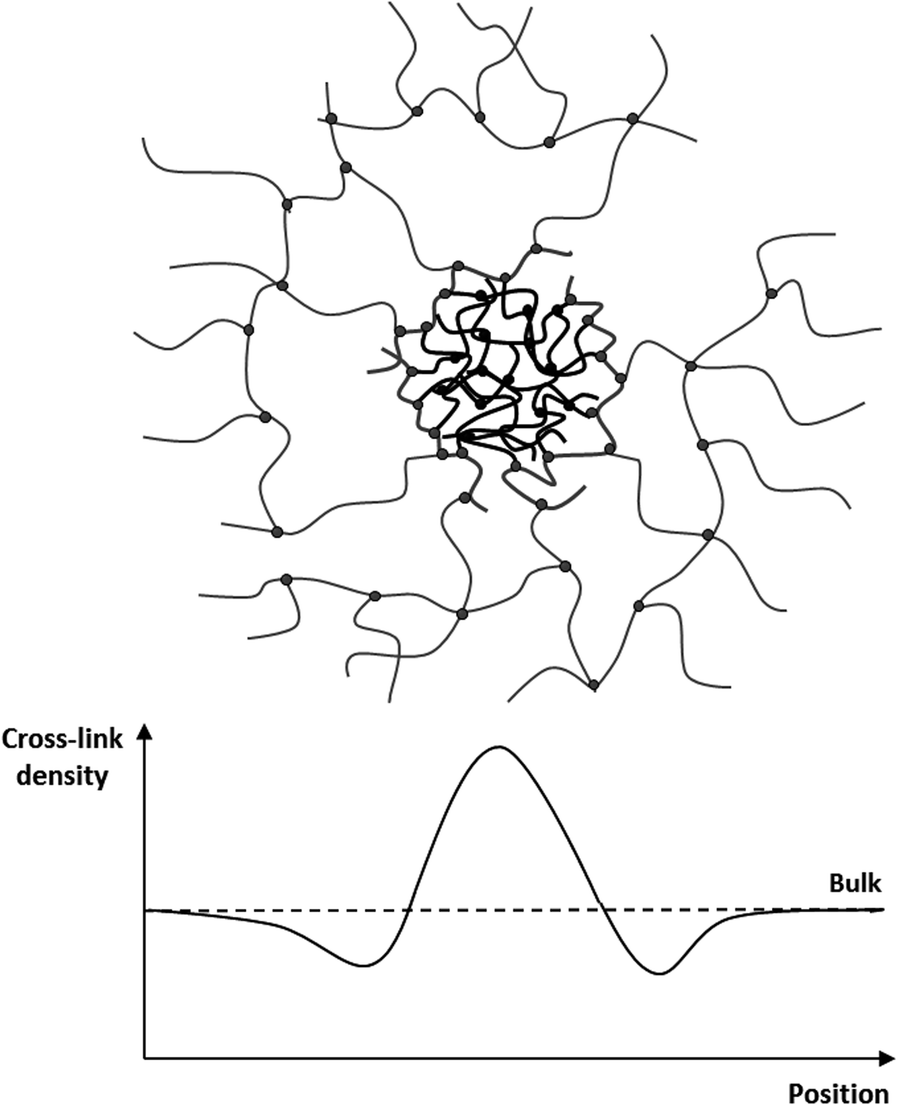 | ||
| Fig. 3 Schematic illustration of the hypothesised efficient cross-linking density. | ||
Interfaces with a lower cross-linking density are still perfectly cross-linked due to the high number of functional groups on each hyperbranched structure, and as such they cannot be regarded as network imperfections, since their mechanical properties are not destroyed.
Very soft silicone elastomer behaviour can also be observed for imperfect networks, which occurs as a result of unreacted groups. This lack of reactivity can be introduced either from stoichiometrically imbalanced reactions27,31 or from incomplete catalyst inhibition reactions.32 It can also arise naturally from steric hindrance or heterogeneity, thus leading to local cross-linker and polymer excesses, respectively. Therefore it is important to ensure that heterogeneous bimodal networks do not contain a large fraction of solubles, so-called sol fractions. The sol fractions of heterogeneous bimodal PDMS networks were determined by conducting swelling experiments with toluene, the results for which can be found in Table S1 in the ESI.† The sol fractions were found to lie in the range of 5–10% and were apparently independent of short chain concentration. The addition of the short chains as domains therefore does not create additional imperfections in the infinite networks, which means that there is complete compatibility between the short-chain domains and the long-chain matrix.
Swelling experiments were performed in order to determine the relationship between the volume degree of equilibrium swelling (Qv) and the mechanical properties. The results can be found in the ESI.†Qv is found to decrease with increasing short-chain concentration, due to the strong increase in cross-link density. Furthermore, as expected, there is not a linear relationship between the elastic moduli (3G′) and the equilibrium swelling degree (see Fig. S3 in the ESI†), which is observed for traditional mono-modal PDMS elastomers. Heterogeneous bimodal networks therefore behave quite different to mono-modal PDMS networks, since the modulus decreases following the addition of short-chain domains, i.e. with increasing cross-link density. This means that swelling capacity is determined from the continuous long-chain part of the network, whereas elasticity is governed by a combination of short- and long-chains properties.
In general, it is difficult to obtain PDMS elastomers with low elastic moduli without deteriorating other network properties. Soft PDMS elastomers therefore usually suffer from high viscous losses. The frequency-dependent viscous loss can be determined from the shear loss modulus G′′. All the prepared bimodal elastomers are seen to have lower viscous loss than the mono-modal PDMS network. Furthermore, the heterogeneous bimodal networks are well cross-linked, as the storage moduli (G′) is 10 to 100 times larger than the loss moduli (G′′) for the investigated frequency range, with differences being smallest at high frequencies. A plot of the storage and loss moduli as functions of frequency can be found as ESI.† Viscous loss at the low strain low frequency limit is measured as a few percentage points only. This is comparable to our previously reported results for heterogeneous bimodal networks.17 Viscous loss in a polymer network is due to dangling chains and sol molecules, i.e. incomplete network formation.27 It is therefore evident from the low viscous losses of the heterogeneous bimodal networks that the obtained low moduli are not a result of incomplete reactions but rather of network artefacts, as shown in Fig. 3. The loops do not contribute significantly to the elastic modulus, but at the same time they do not contribute a loss, since they cannot relax. Heterogeneous bimodal networks are therefore an effective and simple method of creating soft elastomers with very low viscous losses.
The thermal transition behaviour of the heterogeneous bimodal networks was determined using differential scanning calorimetry (DSC). The resulting thermograms can be found as ESI,† while the crystallisation and melting temperatures are summarised in Table S1 in the ESI,† too. A large decrease in the area under the crystallization peaks can be seen in the thermograms, along with an increasing concentration of short-chains. This means that there is a smaller degree of crystallinity in the material in line with increasing short-chain content. Consequently, the PDMS elastomers become inherently amorphous at large concentrations of short chains, as almost no crystalline behaviour is observed. This is most likely due to an increased cross-linking density as the short chain domains are highly dense structures in elastomers. This high cross-link density means that the polymer chains are no longer able to arrange into the positions necessary for crystalline regions to form. Melting temperatures are also observed to decrease with higher concentrations of short chains because of the dilution of the crystalline segments. Furthermore, areas under the melting peaks are reduced at higher concentrations, which mean that the energy required for melting is reduced at larger concentrations of short chains.
Moreover, the effect of short-chain domains on the thermal stability of the PDMS networks was investigated by thermal gravimetric analysis (TGA). The determined degradation temperatures for the bimodal PDMS networks with different short-chain concentrations are summarised in Fig. S5 and Table S1 in the ESI.† The first degradation step occurs due to the cleavage of Si–CH3 bonds. This degradation temperature increases significantly with increasing amounts of short chains (from ∼430 °C to ∼530 °C), which demonstrates that heterogeneous bimodal networks also have a beneficial influence on the thermal stability of PDMS elastomers.
Conclusions
In summary, a novel method of visualising the size of short-chain domains in heterogeneous bimodal PDMS networks was developed. The method allows for the determination of the size of short-chain domains by confocal fluorescence microscopy. The size of the domains was furthermore correlated to the final elastic properties of the bimodal networks for the first time, which makes it possible to explain the surprisingly low elastic moduli obtained for heterogeneous bimodal networks. The results thereby make it possible to tune the properties of PDMS networks in line with the size and concentrations of short-chain hyperbranched structures. This opens up the possibility of heterogeneous bimodal network implementation in advanced applications where strong, unfilled and largely extensible networks are needed, such as in the field of optics and in the emerging field of dielectric elastomers.Acknowledgements
The authors would like to acknowledge Lene Feldskov Nielsen, Coloplast A/S, Humlebæk, Denmark for help with the confocal microscopy and the Danish National Advanced Technology Foundation for its financial support.Notes and references
- J. E. Mark, in Silicon-Based Polymer Science: A Comprehensive Resource, ed. J. M. Zeigler and F. W. G. Fearon, American Chemical Society, 1990, pp. 47–68 Search PubMed.
- C. Ohm, M. Brehmer and R. Zentel, Adv. Mater., 2010, 22, 3366–3387 CrossRef PubMed.
- A. Werber and H. Zappe, Appl. Opt., 2005, 44, 3238–3245 CrossRef.
- J. Ou, T. Glawdel, C. L. Ren and J. Pawliszyn, Lab Chip, 2009, 9, 1926–1932 RSC.
- J. E. Mark, Br. Polym. J., 1985, 17, 144–148 CrossRef CAS.
- G. D. Genesky, B. M. Aguilera-Mercado, D. M. Bhawe, F. a. Escobedo and C. Cohen, Macromolecules, 2008, 41, 8231–8241 CrossRef CAS.
- T. L. Smith, B. Haidar and J. L. Hedrick, Rubber Chem. Technol., 1990, 63, 256–264 CrossRef CAS.
- G. B. Shah and R. W. Winter, Macromol. Chem. Phys., 1996, 197, 2201–2208 CrossRef CAS.
- J. E. Mark, Prog. Polym. Sci., 2003, 28, 1205–1221 CrossRef CAS.
- Z.-M. Zhangt and J. E. Mark, J. Polym. Sci., Polym. Phys. Ed., 1982, 20, 473–480 CrossRef.
- P. Xu and J. E. Mark, Polymer, 1992, 33, 1843–1848 CrossRef CAS.
- M. A. Llorente, A. L. Andrady and J. E. Mark, J. Polym. Sci., Polym. Phys. Ed., 1981, 19, 621–630 CrossRef CAS.
- M. Y. Tang and J. E. Mark, Macromolecules, 1984, 17, 2616–2619 CrossRef CAS.
- B. Viers and J. Mark, J. Macromol. Sci., Part A: Pure Appl. Chem., 2007, 44, 131–138 CrossRef CAS.
- J. E. Mark and A. L. Andrady, Rubber Chem. Technol., 1981, 54, 366–373 CrossRef CAS.
- S.-J. Pan and J. E. Mark, Polym. Bull., 1982, 7, 553–559 CrossRef CAS.
- A. G. Bejenariu, L. Yu and A. L. Skov, Soft Matter, 2012, 8, 3917 RSC.
- F. B. Madsen, I. Dimitrov, A. E. Daugaard, S. Hvilsted and A. L. Skov, Polym. Chem., 2013, 4, 1700–1707 RSC.
- H. Schmid and B. Michel, Macromolecules, 2000, 33, 3042–3049 CrossRef CAS.
- R. Pelrine, R. Kornbluh, Q. Pei and J. Joseph, Science, 2000, 287, 836 CrossRef CAS.
- R. Pelrine, R. Kornbluh and G. Kofod, Adv. Mater., 2000, 12, 1223–1225 CrossRef CAS.
- C. Löwe, X. Zhang and G. Kovacs, Adv. Eng. Mater., 2005, 7, 361–367 CrossRef.
- R. Shankar, T. K. Ghosh and R. J. Spontak, Adv. Mater., 2007, 19, 2218–2223 CrossRef CAS.
- F. B. Madsen, A. E. Daugaard, S. Hvilsted and A. L. Skov, Proc. SPIE, 2013, 8687, 86871H CrossRef PubMed.
- F. B. Madsen, A. E. Daugaard, M. Y. Benslimane, S. Hvilsted and A. L. Skov, Smart Mater. Struct., 2013, 22, 104002 CrossRef.
- F. Chambon and H. H. Winter, J. Rheol., 1987, 31, 683–697 CrossRef CAS.
- S. M. G. Frankær, M. K. Jensen, A. G. Bejenariu and A. L. Skov, Rheol. Acta, 2012, 51, 559–567 CrossRef.
- M. Doi and S. F. Edwards, The Theory of Polymer Dynamics, Oxford University Press, 1999 Search PubMed.
- S. Kunamaneni, D. M. A. Buzza, D. J. Read, D. Parker, A. M. Kenwright, W. J. Feast and A. L. Larsen, Macromolecules, 2006, 39, 6720–6736 CrossRef CAS.
- A. L. Larsen, K. Hansen, P. Sommer-Larsen, O. Hassager, A. Bach, S. Ndoni and M. Jørgensen, Macromolecules, 2003, 36, 10063–10070 CrossRef CAS.
- K. Urayama, T. Miki, T. Takigawa and S. Kohjiya, Chem. Mater., 2004, 16, 173–178 CrossRef CAS.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR spectra and SEC traces of the short-chain hyperbranched structures before and after the reaction, tabulated data from DSC, TGA and swelling experiments, plot of the elastic moduli (3G′) as a function of the degree of equilibrium swelling (Qv), DSC thermograms, TGA curves, fluorescent confocal microscopy images of all samples and plots of the storage and loss moduli as functions of frequency. See DOI: 10.1039/c3ra47522k |
| This journal is © The Royal Society of Chemistry 2014 |