Siloxane-capped amorphous nano-SiOx/graphite with improved dispersion ability and battery anode performance†
Jong-Seon Kim,
Cao Cuong Nguyen,
Hyun-Jin Kim and
Seung-Wan Song*
Dept. of Fine Chemical Engineering & Applied Chemistry, Chungnam National University, Daejeon 305-764, South Korea. E-mail: swsong@cnu.ac.kr; Fax: +82-42-822-6637; Tel: +82-42-821-7008
First published on 24th February 2014
Abstract
Siloxane-capped amorphous nano-SiOx/graphite was prepared by simple reduction and self-organization, and facile particle dispersion. 92% capacity retention of the composite anode is ascribed to effective accommodation of the volume change, and uniform distribution of SiOx on the graphite matrix.
We report simple room temperature preparation and improved performance of amorphous silicon suboxide (SiOx) nanoparticles, whose surface is capped with siloxanes as an artificial solid electrolyte interphase (SEI), and their graphite composite as an anode material for rechargeable lithium batteries. Siloxane-capped amorphous nano-SiO0.26 alone exhibited high, reversible capacities of >2200 mA h g−1 and coulombic efficiencies of 98–99% over 50 cycles at 300 mA g−1 (0.12 C), and excellent rate capability upto 15 A g−1 (6 C) that is superior to previously reported bulk silicon oxides.1–5 The SiO0.26/graphite composite also showed a very stable performance with 92% capacity retention, delivering ∼1000 mA h g−1 and coulombic efficiencies of 99% over 100 cycles at 300 mA g−1 (0.3 C). Hydrophobic surface nature of siloxane-capped nano-SiOx allows a homogeneous dispersion of SiOx nanoparticles on graphite matrix in binder-containing organic solvent (i.e., n-methylpyrrolidinone) just by room temperature dispersion when preparing the slurry. This is advantageous in attaining homogeneous current distribution over the composite electrode and scalable production. Most previous research on silicon-based carbon composites has been conducted including mechanical milling of silicon (Si) or silicon monoxide with carbon materials such as graphite, or carbon-coating on silicon-based materials, followed by high temperature heat treatment typically at higher than 800 °C.1–8 Development of a simple preparation method of the composite and improvement of dispersion ability are yet to be explored.
Si has been considered as a potential alternative anode material to graphite for high-energy rechargeable lithium batteries that are being adopted in advanced mobile electronics, automotive and stationary energy storage systems, due to its high theoretical capacity of 3579 mA h g−1 at room temperature,9 safer operation voltage above lithium and earth-abundance. However, Si suffers from a rapid capacity fade with cycling due to large structural volume change during lithiation–delithiation processes, leading to electrochemical and mechanical disintegration by particle cracking.10 Silicon monoxide has been favored by industry with respect to cycling stability despite sacrificing the capacity and initial coulombic efficiency, as the volume change is better accommodated by the presence of lithium oxide (Li2O) and lithium silicates that form during initial lithiation.3 This study focuses on SiOx (x < 0.5) nanoparticles and their graphite composite, where oxygen and graphite11 can accommodate the volume change. Most of previous SiOx was prepared by plasma treatment or as thin-film form,12 which are not suited for scale-up. Dai et al. showed that chemically prepared crystalline Si/SiOx/SiO2 composite exhibited high capacity retention (>95%) after 350 cycles but low capacity as ∼650 mA h g−1.13 Abel et al. reported that thin film with crystalline Si nanowires with oxygen in both bulk (∼13 at%) and surface delivered improved cycling stability to 80% capacity retention at 300 cycles.12b For higher fraction of oxygen in SiOx, the effect of strain accommodation increases but resistivity increases, so that the oxygen content was suggested to control to less than 0.5.12
Enlarged surface area associated with particle cracking causes intensified interfacial reaction with electrolyte and inhibits surface passivation with a stable SEI. The interfacial cause has been established as the surface attack of LiPF6-derived Lewis acids (PF5, PF3O) and HF (LiPF6 → LiF + PF5; PF5 + H2O → PF3O + 2HF) to Si and their electrochemical reduction, which produce various PF-containing surface species and insulating LiF, and deactivates Si.14 Surface protection such as artificial SEI against the acidic attack is necessary for improving interfacial stability and performance. Herein, we report on facile preparation and improved cycling stability of siloxane-capped amorphous SiOx/graphite composite with homogeneous dispersion as a battery anode. Our report addresses the drawbacks of Si described above and proposes a method of overcoming those issues.
Siloxane-capped amorphous SiOx nanoparticles were first synthesized by reducing the SiCl4 with Li metal, in the presence of methoxytrimethylsilane ((CH3O)Si(CH3)3, MTMS) in room temperature 1,2-dimethoxyethane (glyme) solution in the argon-filled glove box, as illustrated in Scheme 1. Room temperature synthesis can allow the formation of amorphous SiOx by preventing crystal growth, which reduces structural stress associated with volume change in the absence of two-phase regions during lithiation–delithiation.15 Although syntheses of Si from SiCl4 (ref. 16) and multi-step surface capping with relatively long chain alkyl group16b,c,17 had been previously conducted, they focused only on crystalline Si. Thus thermal treatment was conducted. The role of MTMS is to form stable Si–O–Si siloxanes at the surface of SiOx by self-organized condensation between methoxy group of silane and surface Si–OH or Si–O group of SiOx14 and/or to partly act as an oxygen source to form Si–O bond. After removing side products and residual glyme followed by washing with acetone–water solution and vacuum drying, we finally obtained fine dark grey powders (Fig. S1†) with the chemical formula of SiO0.26±0.02, which is determined by HRTEM mapping. X-ray diffraction (XRD) pattern (Fig. 1A) shows very broad features, characteristic of amorphous material. IR spectrum (Fig. 1B) shows a dominant and broad feature at 1060 cm−1, attributed to νasym(Si–O–Si) from amorphous SiO0.26 with surface siloxane, respectively.18 Peaks at 1251 and 841 cm−1 are due to δ(Si–CH3), ν(Si–C) and δ(CH3) of trimethyl-terminated siloxane.18 Tiny peak near 2957 cm−1 due to –CH3 group supports this assignment. IR data indicate that the surface of amorphous SiO0.26 particles is capped by trimethyl-terminated siloxanes. AC Impedance spectral analysis results confirmed the presence of surface siloxanes (Fig. S5†). Relative ratio of siloxanes/SiO0.26 was 12/88 wt% determined using the carbon content of trimethyl groups by automatic elemental analysis. The pictures in Fig. 1C and D and S1† demonstrate an excellent dispersion ability of siloxane-capped amorphous SiO0.26 particles in organic media, due to their hydrophobic surface. The estimated size of a primary particle is in the range of 10–25 nm in diameter and BET surface area measured (Fig. S2†) is 170 m2 g−1. Dispersed feature of selected area electron diffraction (SAED) pattern and the absence of lattice fringes in the high resolution (HR) TEM image (Fig. 1E) confirm that the SiO0.26 is amorphous.
 | ||
| Scheme 1 Synthesis of siloxane-capped amorphous SiOx nanoparticles. | ||
 | ||
| Fig. 1 (A) XRD pattern, (B) ATR FTIR spectrum, (C) high dispersion ability in acetone, and (D and E) HR-TEM images and SAED pattern of siloxane-capped amorphous nano-SiO0.26. | ||
Electrochemical charge (lithiation)–discharge (delithiation) cycling ability of siloxane-capped amorphous nano-SiO0.26 electrode (Fig. 2A–D) was tested in coin-cells in the electrolyte of 1 M LiPF6/fluoroethylene carbonate (FEC)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :diethyl carbonate (DEC) (1:1 volume ratio, Panax E-Tec) with 3 wt% trimethyl phosphite ((CH3O)3P) as an acid (e.g., PF5, PF3O)-scavenging additive,19 in the voltage range of 0.01 and 1.5 V at a constant current (300 mA g−1, ∼0.12 C rate)–constant voltage (0.01 V) mode. Nano-Si electrode in FEC-based electrolyte is noticed to outperform the one in conventional ethylene carbonate (EC)-based electrolyte that tends to show inferior SEI stability and performance (Fig. S3†).15,20 The gravimetric specific capacities were normalized by the mass of SiO0.26. The electrode (Fig. 2A) delivers the first charge and discharge capacities of 3458 and 2481 mA h g−1, respectively, resulting in initial coulombic efficiency of 72%. This is in contrast to low initial coulombic efficiency below 65% for SiOx (x > 0.5).14c,15 Coulombic efficiency increases to higher than 98% after the fifth cycle. In the first cycle of differential capacity plot (Fig. 2B), little cathodic capacity above 0.65 V indicates that electrolyte reduction is negligible at that relatively high voltage region. This implies that the presence of oxygen, surface capping with siloxanes and the use of FEC-based electrolyte are effective in suppressing initial electrolyte reduction. Tiny cathodic peak at 0.35 V is attributed to the reaction of SiO0.26 with lithium forming Li2O, lithium silicates and Si. Subsequent large peaks at 0.18 and 0.07 V are due to lithiation of amorphous Si forming LiySi. In the reverse process, anodic peaks at 0.26 and 0.45 V are by delithiation from LiySi regenerating the amorphous Si.9 Structural resolution of prominent peaks by lithiation–delithiation is preserved till the 50th cycle, demonstrating a high reaction reversibility.
:diethyl carbonate (DEC) (1:1 volume ratio, Panax E-Tec) with 3 wt% trimethyl phosphite ((CH3O)3P) as an acid (e.g., PF5, PF3O)-scavenging additive,19 in the voltage range of 0.01 and 1.5 V at a constant current (300 mA g−1, ∼0.12 C rate)–constant voltage (0.01 V) mode. Nano-Si electrode in FEC-based electrolyte is noticed to outperform the one in conventional ethylene carbonate (EC)-based electrolyte that tends to show inferior SEI stability and performance (Fig. S3†).15,20 The gravimetric specific capacities were normalized by the mass of SiO0.26. The electrode (Fig. 2A) delivers the first charge and discharge capacities of 3458 and 2481 mA h g−1, respectively, resulting in initial coulombic efficiency of 72%. This is in contrast to low initial coulombic efficiency below 65% for SiOx (x > 0.5).14c,15 Coulombic efficiency increases to higher than 98% after the fifth cycle. In the first cycle of differential capacity plot (Fig. 2B), little cathodic capacity above 0.65 V indicates that electrolyte reduction is negligible at that relatively high voltage region. This implies that the presence of oxygen, surface capping with siloxanes and the use of FEC-based electrolyte are effective in suppressing initial electrolyte reduction. Tiny cathodic peak at 0.35 V is attributed to the reaction of SiO0.26 with lithium forming Li2O, lithium silicates and Si. Subsequent large peaks at 0.18 and 0.07 V are due to lithiation of amorphous Si forming LiySi. In the reverse process, anodic peaks at 0.26 and 0.45 V are by delithiation from LiySi regenerating the amorphous Si.9 Structural resolution of prominent peaks by lithiation–delithiation is preserved till the 50th cycle, demonstrating a high reaction reversibility.
 | ||
| Fig. 2 (A) Voltage profiles, and plots of (B) differential capacity and (C) discharge capacity and coulombic efficiency of siloxane-capped amorphous nano-SiO0.26 electrode, and (D) high-rate capability at the rate from 0.12 (300 mA g−1) to 6 C (15 A g−1). | ||
The electrode exhibits a stable cycling performance with capacity retention of 89% of the first discharge capacity at the 50th cycle (Fig. 2C), delivering discharge capacities of 2481–2206 mA h g−1 and high coulombic efficiencies 98–99%. The capacity and cycling stability, even in the absence of carbon-coating, are superior to previously reported silicon oxide-based electrodes (also see Fig. S4†).2–5,12 Enlargement of particle size or particle cracking, which was usually observed on cycled Si-based anodes, are not observed in our electrode (Fig. S6†). Excellent rate capability is obtained (Fig. 2D); the capacity of 1673 mA h g−1 at 6 C (15 A g−1) is 67% of the one at 0.12 C. We believe that such superior cycling stability and rate performance are ascribed to nanosize and amorphous characteristics of SiO0.26, homogeneous dispersion with carbon black powders and improved interfacial stability, which lead to reduced structural and mechanical strain upon volume change, and fast Li+-diffusion and electron transport kinetics.
Particles of artificial SEI-capped nano-SiO0.26 and graphite matrix in an equal weight ratio are homogenously dispersed over composite electrode, as illustrated in Fig. 3A and revealed in scanning electron microscopic (SEM) and elemental mapping images of Fig. 3B.
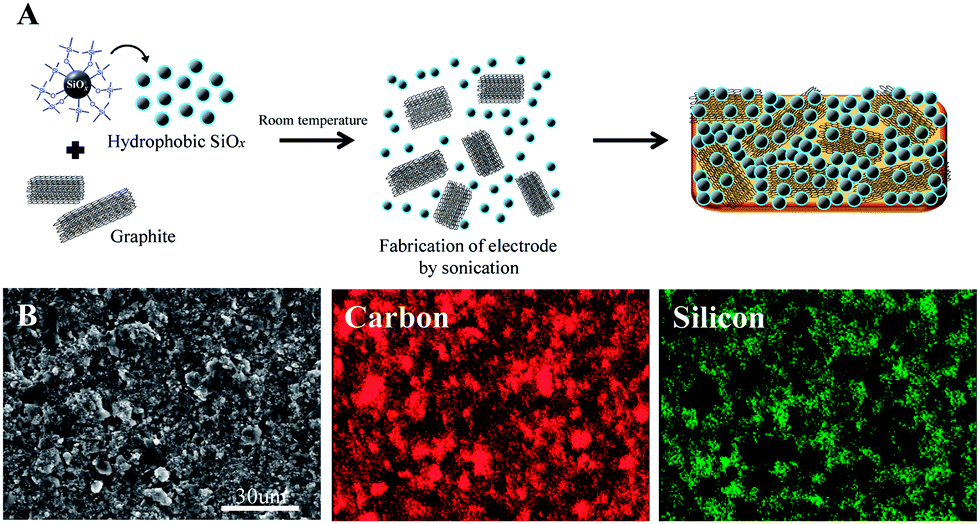 | ||
| Fig. 3 (A) Facile preparation of SiO0.26/graphite electrode and (B) its SEM elemental mapping images. | ||
Cycling performance of SiO0.26/graphite composite electrode at the rate of 0.3 C (300 mA g−1) in 1 M LiPF6/FEC:DEC with 3 wt% trimethyl phosphite is displayed in Fig. 4. Initial charge and discharge capacities (Fig. 4A) are 1370 and 1010 mA h g−1, respectively, which are determined based on the total weight of SiO0.26/graphite composite, and initial coulombic efficiency of 74%. The electrode delivers discharge capacities of 1010–931 mA h g−1 with the outstanding capacity retention of 92% at the 100th cycle and sustained coulombic efficiencies of 99%. This capacity is over 2.5 times higher than the theoretical capacity of graphite and can increase further by lowering the applied current from 300 mA g−1, which is somewhat high current close to 0.8 C for graphite. It is surprising to see such outstanding cycling stability just by a simple dispersion of two materials in room temperature binder solution, in contrast to inevitable ball-milling followed by high-temperature thermal treatment for obtaining improved capacity retention of Si/graphite and SiO/graphite composite anodes.1–5 The SEM imaging after cycling reveals a sustained robust microstructure without particle cracking or separation (Fig. S7†). The presence of graphite matrix and siloxanes at the surface of nano-SiO0.26 and their homogeneous dispersion are synergistically effective in accommodating the volume change, and providing interfacial stability, microstructural robustness and finally cycling stability.
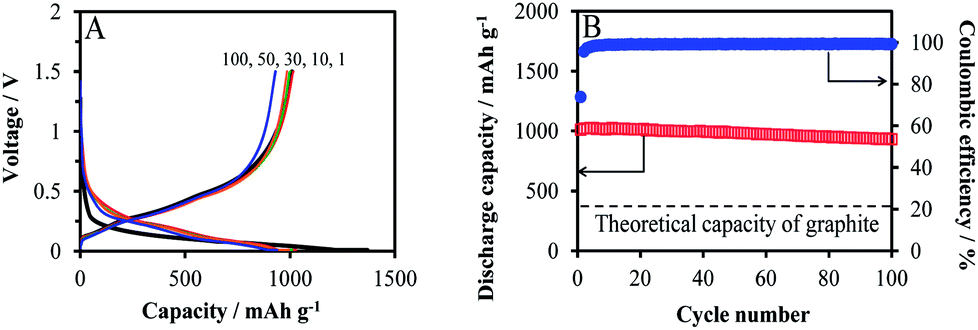 | ||
| Fig. 4 (A) Voltage profiles and (B) cycling performance of SiO0.26/graphite composite electrode at 0.3 C (300 mA g−1). | ||
In conclusion, we have demonstrated a simple and facile room temperature preparation of siloxane-capped amorphous SiOx nanoparticles and its graphite composite. The outstanding cycling stability of SiO0.26/graphite composite electrode is ascribed to structural and interfacial stabilization, and improved particle dispersion ability and electrical conductivity. The concept presented here is potentially applicable to other electrode materials, which suffer from structural stress, interfacial instability to electrolyte and particle dispersion of different components, for not only lithium-ion batteries but also various other batteries.
Acknowledgements
This work was supported by the Ministry of Education (2012026203).Notes and references
- X. Yang, P. Zhang, C. Shi and Z. Wen, Electrochem. Solid-State Lett., 2012, 1, M5 CrossRef CAS PubMed.
- J.-H. Kim, H.-J. Sohn, H. Kim, G. Jeong and W. Choi, J. Power Sources, 2007, 170, 456 CrossRef CAS PubMed.
- Y. Nagao, H. Sakaguchi, H. Honda, T. Fukunaga and T. Seaka, J. Electrochem. Soc., 2004, 151, A1572 CrossRef CAS PubMed.
- J. Yang, Y. Takeda, N. Imanishi, C. Capiglia, J. Y. Xie and O. Yamamoto, Solid State Ionics, 2002, 152–153, 125 CrossRef CAS.
- F. Dai, R. Yi, M. L. Gordin, S. Chen and D. Wang, RSC Adv., 2012, 2, 12710 RSC.
- M. K. Datta and P. N. Kumta, J. Power Sources, 2006, 158, 557 CrossRef CAS PubMed.
- P. Zuo, G. Yin and Y. Ma, Electrochim. Acta, 2007, 52, 4878 CrossRef CAS PubMed.
- Z. P. Guo, E. Milin, J. Z. Wang, J. Chen and H. K. Liu, J. Electrochem. Soc., 2005, 152, A2211 CrossRef PubMed.
- (a) W. J. Weydanz, M. Wohlfhart-Mehrens and R. A. Huggins, J. Power Sources, 1999, 237, 81–82 Search PubMed; (b) M. N. Obrovac and L. Christensen, Electrochem. Solid-State Lett., 2004, 7, A93 CrossRef CAS PubMed.
- (a) L. Y. Beaulieu, K. W. Wberman, R. L. Turner, L. J. Krause and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, 137 CrossRef PubMed; (b) M. N. Obrovac and L. J. Krause, J. Electrochem. Soc., 2007, 154, A103 CrossRef CAS PubMed; (c) J. H. Ryu, J. W. Kim, Y.-E. Sung and S. M. Oh, Electrochem. Solid-State Lett., 2004, 7, A306 CrossRef CAS PubMed.
- X.-Q. Yang, J. McBreen, W.-S. Yoon, M. Yoshio, H. Wang, K. Fukuda and T. Umeno, Electrochem. Commun., 2002, 4, 893 CrossRef CAS.
- (a) K. Kim, J.-H. Park, S.-G. Doo and T. Kim, Thin Solid Films, 2010, 518, 6547 CrossRef CAS PubMed; (b) P. R. Abel, Y.-M. Lin, H. Celio, A. Heller and C. B. Mullins, ACS Nano, 2012, 6, 2506 CrossRef CAS PubMed.
- F. Dai, R. Yi, M. L. Gordin, S. Chen and D. Wang, RSC Adv., 2012, 2, 12710 RSC.
- (a) S.-W. Song and S.-W. Baek, Electrochem. Solid-State Lett., 2009, 12, A23 CrossRef CAS PubMed; (b) C. C. Nguyen and S.-W. Song, Electrochim. Acta, 2010, 55, 3026 CrossRef CAS PubMed; (c) Y.-G. Ryu, S. S. Lee, S.-K. Mah, D.-J. Lee, K. Kwon, S.-S. Hwang and S.-G. Doo, J. Electrochem. Soc., 2008, 155, A583 CrossRef CAS PubMed.
- (a) T. D. Hatchard and J. R. Dahn, J. Electrochem. Soc., 2004, 151, A838 CrossRef CAS PubMed; (b) L. Y. Beaulieu, K. C. Hewitt, R. L. Turner, A. Bonakdarpour, A. A. Abdo, L. Christensen, K. W. Eberman, L. J. Krause and J. R. Dahn, J. Electrochem. Soc., 2003, 150, A149 CrossRef CAS PubMed; (c) C. C. Nguyen, H. Choi and S.-W. Song, J. Electrochem. Soc., 2013, 160, A906 CrossRef CAS PubMed.
- (a) J. R. Heath, Science, 1992, 258, 1131 CAS; (b) R. K. Baldwin, K. A. Pettigrew, J. C. Garno, P. P. Power, G.-Y. Liu and S. M. Kauzlarich, J. Am. Chem. Soc., 2002, 124, 1150 CrossRef CAS PubMed; (c) R. K. Baldwin, K. A. Pettigrew, E. Ratai, M. P. Augustine and S. M. Kauzlarich, Chem. Commun., 2002, 1822 RSC; (d) A. Kornowski, M. Giersig, R. Vogel, A. Chemseddine and H. Weller, Adv. Mater., 1993, 5, 634 CrossRef CAS; (e) N. Dhas, C. P. Raj and A. Gedanken, Chem. Mater., 1998, 10, 3278 CrossRef CAS.
- J. Zou, R. K. Baldwin, K. A. Pettigrew and S. M. Kauzlarich, Nano Lett., 2004, 4, 1181 CrossRef CAS.
- (a) G. Socrates, Infrared Characteristic Group Frequencies: Tables and Charts, John Wiley & Sons, New York, 2nd edn, 1994 Search PubMed; (b) Spectral database for organic compounds SDBS, http://riodb01.ibase.aist.go.jp/sdbs.
- M.-H. Choo, C. C. Nguyen, S. Hong, Y. H. Kwon, S.-W. Woo, J. Y. Kim and S.-W. Song, Electrochim. Acta, 2013, 112, 252 CrossRef CAS PubMed.
- (a) H. Nakai, T. Kubota, A. Kita and A. Kawashima, J. Electrochem. Soc., 2011, 158, A798 CrossRef CAS PubMed; (b) Y.-M. Lin, K. C. Klavetter, P. R. Abel, N. C. Davy, J. L. Snider, A. Heller and C. B. Mullins, Chem. Commun., 2012, 48, 7268 RSC; (c) Y. Oumella, D. Delpuech, D. Mazouzi, N. Dupré, J. Gaubicher, P. Moreau, P. Soudan, B. Lestries and D. Guyomard, J. Mater. Chem., 2011, 21, 6201 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47476c |
| This journal is © The Royal Society of Chemistry 2014 |