DOI:
10.1039/C3RA45702H
(Review Article)
RSC Adv., 2014,
4, 12882-12917
meta-Chloroperbenzoic acid (mCPBA): a versatile reagent in organic synthesis
Received
14th October 2013
, Accepted 16th January 2014
First published on 17th January 2014
Abstract
The synthetic uses of different peroxides for organic synthesis have been widely studied. Among these peroxides, meta-chloroperbenzoic acid (mCPBA) is an efficient oxidizing reagent and have been used for many oxidative transformations. mCPBA is widely used for chemical transformations such as the oxidation of carbonyl compounds, iminoindolines, olefins, imines, alkanes, silyl enol ethers, N- and S-heterocycles, active methylene groups, fluoromethylated allylic bromides, cyclic acetals, N-substituted phthalimidines, selenides, furans and phosphates. The purpose of this review is to collect and discuss the synthetic applications of meta-chloroperbenzoic acid (mCPBA) over the past few decades.
 Hidayat Hussain | Dr Hidayat Hussain received his PhD in 2004 from HEJRIC, Pakistan. From Jun. 2004 to Sept. 2007 he was a postdoctoral fellow at the University of Paderborn, Germany. After finishing a one year postdoctoral fellowship (from Oct. 2007 to Nov. 2008) at the University of Maine, France, he returned in December 2008 to the University of Paderborn as a senior postdoctoral associate and worked there until Oct. 2010. He currently works at the UoN Chair of Oman's Medicinal Plants and Marine Natural Products, University of Nizwa, Oman. Some of his research interests include the synthesis of bioactive molecules and the discovery of new therapeutic agents from fungi and plants. To date he has authored and co-authored over 145 scientific publications with a cumulative impact factor of ca. 300 and he is a referee for 20 international journals. |
 Ahmed Al-Harrasi | Dr Ahmed Al-Harrasi received his PhD in Organic Chemistry in 2005 from Free University of Berlin as a DAAD-fellow under the supervision of Prof. Hans-Ulrich Reissig. His PhD work was on New Transformations of Enantiopure 3,6-dihydro-2H-1,2-oxazines. He then worked as a postdoctoral Fulbright-fellow in 2008 under the supervision of Prof. Tadhg Begely on the topic “synthesis of isotopically-labeled thiamin pyrophosphates”. His current research focuses on drugs discovered from Omani medicinal plants and marine species as well as on the synthesis of biologically-active compounds. He is currently the chairperson of the Chair of Oman's Medicinal Plants and Marine Natural Products and Assistant Dean for Graduate Studies and Research at the University of Nizwa, Oman. He has several funded projects with a budget that exceeds two million USD. He has authored and co-authored over 75 scientific papers. |
 Ivan R. Green | I. R. Green graduated with a PhD in Organic Chemistry in 1973 from University of Cape Town. He was made a full Professor in 1986 and a Senior Professor in 1990 at the University of the Western Cape where he lectured for 39 years until his retirement in July 2011.To date, he has authored and co-authored over 120 scientific publications, given 40 podium lectures at international conferences and has supervised 30 MSc and 18 PhD students locally and 6 PhD students internationally. He is a referee for 8 international journals. Upon retirement he moved to the University of Stellenbosch where he is involved in mentoring research students, gives seminars and is involved in alkaloid research. |
 Ishtiaq Ahmed | Dr Ishtiaq Ahmed received his PhD in 2007 under the supervision of Prof. Karsten Krohn with a thesis on the study of enantioselective epoxidation, asymmetric reduction and the synthesis of bioactive oligomeric flavonoids. He carried out his postdoctoral research in the same group working on the synthesis of chiral macrolide building blocks from sugars, the synthesis of anthrapyran antibiotics, isolation and the structure elucidation of secondary metabolites from fungi and medicinal plants. Dr Ishtiaq moved to the DFG-Centre for Functional Nanostructures, Karlsruhe Institute of Technology (KIT) in November 2010 as a postdoctoral researcher. He is currently working on the design of photoswitchable, multifunctional linkers for nanoparticles, DNA and protein modification under the supervision of Dr Ljiljana Fruk. |
 Ghulam Abbas | Dr Abbas completed his PhD at HEJRIC, Pakistan. During his PhD studies, he was actively involved in many research oriented activities and won many poster awards at international conferences and symposia. He received training at The Rockefeller University New York, USA for 14 months while working on diabetes and late diabetic complications at the molecular level. Dr Abbas has published 15 research articles in international journals. He is currently involved in establishing new standard bio-assays to understand various diseases (particularly diabetes and late diabetic complications) at the molecular level and to discover new non-toxic lead molecules of Natural and Synthetic origin in order to inhibit these diseases and to find out the mechanism of action of new potent molecules. He currently works for the UoN Chair of Oman's Medicinal Plants and Marine Natural Products at the University of Nizwa, Oman. |
 Najeeb Ur Rehman | Dr Najeeb Ur Rehman received his M.Sc degree in 2005 and PhD in 2012 from Kohat University of Science & Technology, Kohat, Pakistan. His PhD topic was on the isolation, structure elucidation and biological activities of Nepeta clarkei and related species. He is an author/co-author of more than 30 national and international publications. Currently, he is working as a postdoctoral fellow at the UoN Chair of Oman's Medicinal Plants and Marine Natural Products, University of Nizwa, Oman. He is also a member of the Phytochemical Society of Europe. |
1. Introduction
Oxidation reactions in synthetic organic chemistry constitute one of the more important transformations employed on a regular basis and are widely used in the production of pharmaceuticals, agrochemicals and their intermediates.1–6 However, oxidations are among the most problematic processes in terms of their safety, environmental friendliness and operational simplicity. Often the severe reaction conditions as well as the highly reactive nature of the oxidants involved restrict their application to large scale synthesis protocols, which is most likely a reason why so many basic research papers and new patents dealing with such fundamental transformations have appeared in the recent past.6
Organic peroxides, because of their exceptional reactivity and oxidative potential are widely used in research laboratories. Organic peracids are versatile reagents capable of oxidizing a variety of functional groups under generally mild conditions. Among these organic peroxides, meta-chloroperbenzoic acid (mCPBA) is a peroxycarboxylic acid used widely as an oxidant in organic synthesis due to its versatile oxidizing power and relative ease of handling.7 Its unique reactivity is characterized by a weak O–O bond and a nucleophilic OH group. The O–O bond of mCPBA transfers an oxygen atom to electron-rich substrates, while the nucleophilic attack of mCPBA on ketones and aldehydes results in the insertion of an oxygen atom.8
Initially, mCPBA was used extensively for determination of the total unsaturation in various types of organic compounds.9 In recent decades, mCPBA has been used in the oxidation of carbonyl compounds, alcohols, iminoindolines, olefins, alkynes, carboxylic acids, amines, imines, alkanes, silyl enol ethers, N- and S-heterocycles, active methylene groups, fluoromethylated allylic bromides, cyclic acetals, ketals, diazoketones, N-substituted phthalimidines, selenides, furans, phosphates, and for N-oxidation.10 mCPBA can be prepared by the reaction of m-chlorobenzoyl chloride with H2O2 in the presence of MgSO4·7H2O, aqueous NaOH and dioxane (Scheme 1).11
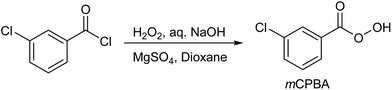 |
| | Scheme 1 Preparation of mCPBA. | |
mCPBA is easy to handle, flammable, and hygroscopic, and pure mCPBA is shock-sensitive and can deflagrate. Moreover, it is potentially explosive beyond 85% purity and exhibits 1% degradation per year at room temperature.10 It is interesting to note that 85% mCPBA is not shock-sensitive, but it should be stored in a refrigerator in a tightly closed container. mCPBA irritates the mucous membranes, respiratory tract, eyes and skin. Moreover, skin contact with mCPBA causes burns and blisters. Therefore, it is recommended that mCPBA should only be used in a chemical fume hood.
mCPBA has been frequently employed over the years with many examples of its use on pilot scale and pharmaceutical manufacturing.12 However, safety concerns related to its use on scale-up are also well-known, with the pure solid being shock-sensitive and potentially explosive in the condensed phase.12 Commercial grade (70–77 wt%) mCPBA, although somewhat stabilised with chlorobenzoic acid and water, still represents a significant concern when used on a large scale.12 It has been reported that CH2Cl2 could be a safer solvent for the preparation of mCPBA solutions, but it has recently been reported that CH2Cl2 cannot be viewed as an inherently safer solvent for preparation of mCPBA solutions at high concentrations (large scale). However, upon implementation of some safety measures, mCPBA/DMF solutions could be successfully applied on a large scale. mCPBA is a white powder and soluble in CH2Cl2, CHCl3, 1,2-dichloroethane, ethylacetate, benzene, and ether. However mCPBA is slightly soluble in hexane and insoluble in water.13
In this review, we summarise the most important accomplishments in the chemistry of mCPBA-mediated oxidative transformations, with the hope of encouraging the development of more novel, prospective synthetic applications in the near future. The review will abbreviate meta-chloroperbenzoic acid as mCPBA in the subsequent sections.
2. Named reactions
2.1. The Baeyer–Villiger oxidation
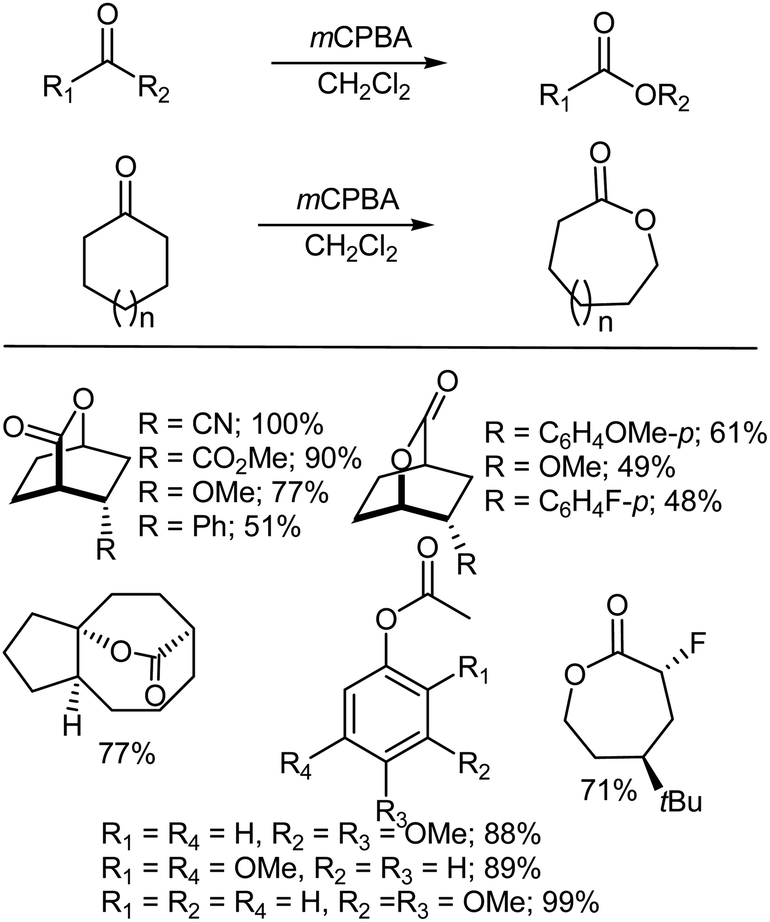
The Baeyer–Villiger oxidation represents an important process for the synthesis of lactones and esters from ketones.14–21 The reaction is of great importance for the manufacture of lactones (Scheme 2). The regiospecificity of the reaction depends on the relative migratory ability of the substituents attached to either side of the carbonyl group. In general, it has been found that substituents which are able to stabilize a positive charge migrate more readily, which has lead to the establishment of an order of preference viz., tert-alkyl > cyclohexyl > sec-alkyl > phenyl > primary-alkyl > CH3. In some cases, stereoelectronic or ring strain factors also affect the regiochemical outcome. The reaction of aldehydes preferably gives formates, but sometimes only the liberated alcohol may be isolated due to the solvolytic instability of the product formate under the reaction conditions.15,22,23 The Baeyer–Villiger oxidation with a peroxyacid involves the nucleophilic addition of the peroxide reagent onto the carbonyl carbon of the substrate to afford a tetrahedral Criegee intermediate.24 The intermediate undergoes the intramolecular rearrangement of an alkyl or aryl substituent from the central carbon to the adjacent oxygen and this migration is accompanied by cleavage of the weak O–O bond and simultaneous formation of the ester (or lactone) and a carboxylic acid22 (Scheme 2).
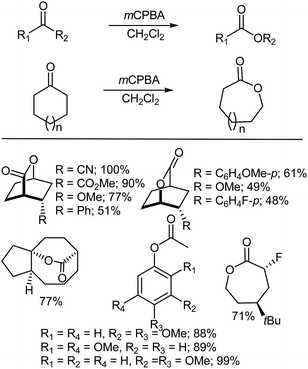 |
| | Scheme 2 Examples of the Baeyer–Villiger oxidation of ketones using mCPBA. | |
Feng et al.25 reported on the catalytic enantioselective Baeyer–Villiger oxidations of racemic and meso cyclic ketones in the presence of chiral N,N′-dioxide–ScIII complex catalysts. The asymmetric Baeyer–Villiger oxidation of racemic or prochiral cyclic ketones provides a simple and attractive route for the synthesis of optically active lactones.26–28 Various meso-cyclohexanones provided the corresponding ε-lactones with excellent enantioselectivities (Scheme 3). The enantiocontrol of the reaction was sensitive to neither the electronic properties nor the steric hindrance of substituents on the phenyl ring of 4-aryl-substituted cyclohexanones. Generally, the desired chiral 5-aryl-substituted ε-lactones were isolated with excellent enantioselectivities (up to 95% ee) and in good yields (up to 90%). Moreover, fused-ring-substituted cyclohexanones were also tolerated, giving the desired products with excellent ee values. Catalytic systems for the Baeyer–Villiger oxidation of a variety of meso-cyclobutanones were also explored, and the desired γ-lactones were obtained in good yields (up to 99%) with good enantioselectivities (up to 91% ee) (Scheme 3). It is interesting to note that the electronic nature of substituents in the cyclobutanone starting materials had almost no effect on the efficiency and enantioselectivity of the reaction.25
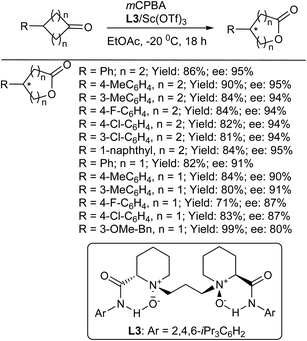 |
| | Scheme 3 Substrate scope for the desymmetrization of meso-cyclohexanones. | |
With regard to the kinetic resolution of racemic cyclic ketones through the Baeyer–Villiger oxidation, the stereochemistry is affected not only by stereoelectronic control, but also by chiral recognition. The first examples of the Baeyer–Villiger oxidation of racemic cyclic ketones were independently reported by the groups of Bolm29 and Strukul.30 In general, the normal CHR group-migrated product [i.e., the “normal” lactone (NL)] distribution, which depends on the migratory aptitude (tertiary > secondary > primary), was observed. Feng et al.25 reported on the kinetic resolution of racemic cyclic ketones using the Baeyer–Villiger oxidation in the presence of a chiral N,N′-dioxide–ScIII complex catalyst. A series of optically active ε- and γ-lactones were obtained with excellent outcomes. The latter reaction using the ScIII catalyst, in particular, gave the “abnormal” lactone (AL) derived from the preferential migration of a CH2 group with high enantioselectivity (Table 1). The kinetic resolution of a series of racemic 2-aryl-cyclohexanones 1 was then specifically examined. Interestingly, compounds AL-2a–f were obtained as the major products. Generally, the lactones AL-(R)-2 and unreacted ketones (S)-1 were isolated with high conversion, ee values and good to excellent AL-(R)-2/NL-3 ratios (from 5.6/1 to >19/1). The reaction efficiency and enantiocontrol were sensitive to the electronic properties of the substituent on the phenyl nucleus of the substrate. Substrates with an electron-withdrawing substituent (e.g., Cl, Br) gave the unreacted ketone (S)-1 and corresponding lactone AL-(R)-2 with higher ee values than those with electron-donating one (e.g., Me, OMe). Moreover, 2-substituted fused ring ketones 1 were also tolerated, giving the products and unreacted ketones with good ee values.25
Table 1 Substrate scope for the kinetic resolution of racemic 2-aryl-substituted cyclohexanones
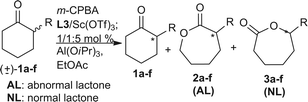
|
| |
Yield (%) |
ee |
| R |
1 |
2 |
1 |
2 + 3 |
2/3 |
| Ph |
94 (S) |
94 (S) |
46 |
51 |
17/1 |
| 4-ClC6H4 |
97 (S) |
93 (S) |
47 |
53 |
19/1 |
| 4-BrC6H4 |
97 (S) |
92 (S) |
47 |
53 |
19/1 |
| 4-MeC6H4 |
89 (S) |
91 (S) |
51 |
48 |
12/1 |
| 4-MeOC6H4 |
82 (S) |
90 (S) |
44 |
56 |
6/1 |
| 1-Naphthyl |
99 |
98 |
49 |
51 |
19/1 |
2.2. Meisenheimer rearrangement
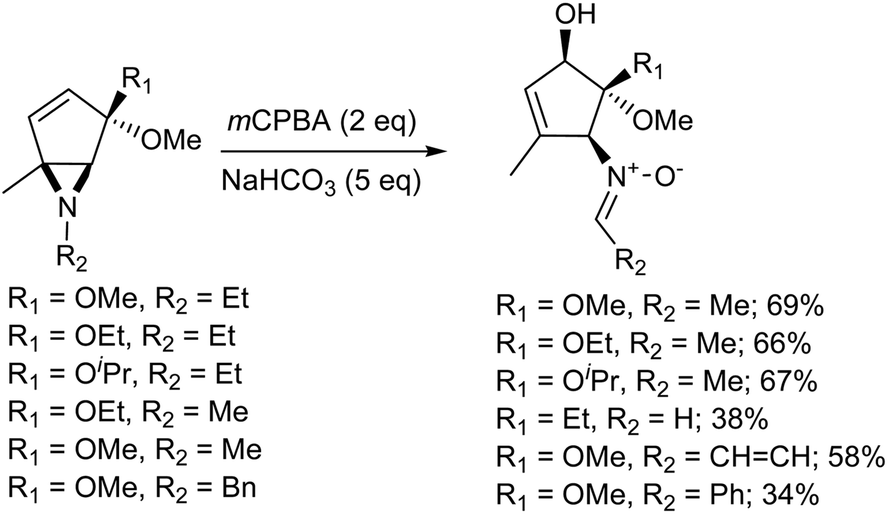
The Meisenheimer rearrangement is perhaps of more interest from a synthetic point of view, as it allows the transfer of both functionality and stereochemical information. Penkett and Simpson31 reported the oxidation of a range of aziridines with mCPBA, which gave products arising from the [2,3]-Meisenheimer rearrangement of the initial N-oxides, followed by a further oxidation to give nitrones. It was found that two equivalents of mCPBA were required. It was also found that the reaction gave low yields of product in CH2Cl2, but that the yield improved considerably when the reaction was carried out in methanol or acetonitrile.
For good selectivity to be observed, it is necessary for the oxidation of the aziridine to be faster than the oxidation of the nitrone. The initial oxidation step is likely to involve a larger increase in dipole moment than the second and thus a polar solvent such as acetonitrile would be expected to improve the selectivity towards the nitrone. Methanol is also a very polar solvent. However the aziridinyl nitrogen lone pair of the aziridine would, to an extent, be deactivated towards oxidation by hydrogen bonding to the solvent. This would reduce the rate of the initial oxidation and perhaps account for the reversal of selectivity. It seems likely that the reaction proceeds via the oxidation of the aziridine give the N-oxide, which undergoes a rapid Meisenheimer rearrangement followed by the further oxidation of the nucleophilic nitrogen, and then base catalysed N–O bond cleavage gives the nitrone (Scheme 4).31
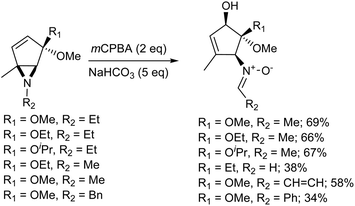 |
| | Scheme 4 The oxidation of aziridines. | |
2.3. Cope elimination
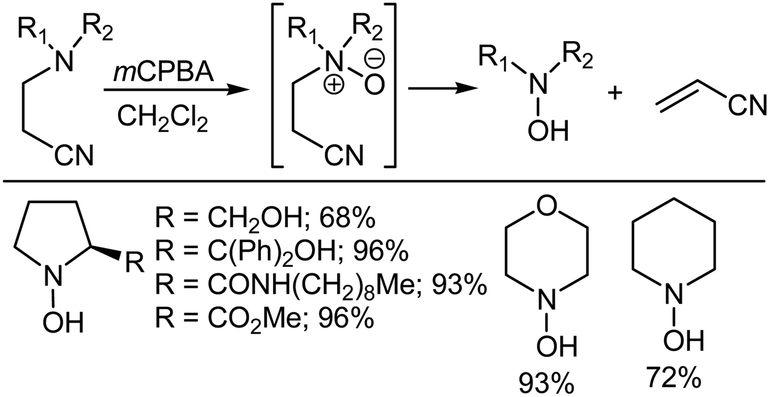
Nagasawa et al.32 first reported that the use of an appropriately positioned β-electron withdrawing nitrile group allows the Cope elimination to occur at significantly lower temperatures than usually required. In another study, O'Neil et al.33 reported the oxidation of a range of β-cyanoethyl tertiary amines using mCPBA to afford the corresponding N-oxides, which could either be isolated or allowed to undergo Cope elimination to give secondary hydroxylamines (Scheme 5). The reaction works for both cyclic and acyclic systems.
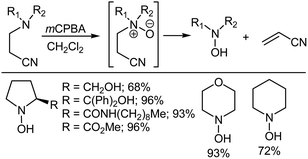 |
| | Scheme 5 The synthesis of secondary hydroxylamines via the Cope elimination of (β-cyanoethyl)amines. | |
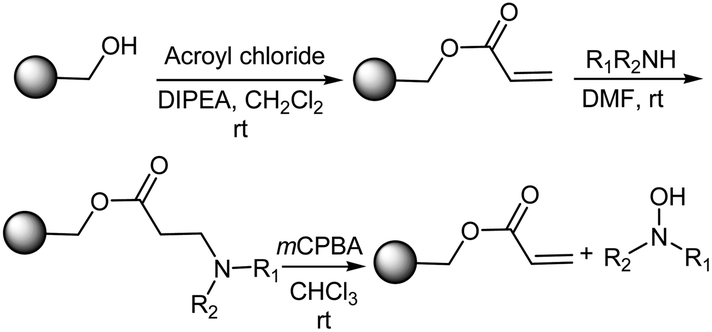
Sammelson and Kurth34 reported a solid phase oxidation–Cope elimination for the synthesis of N,N-disubstituted hydroxylamines from REM resin (polymer-bound benzyl acrylate) (Scheme 6). This solid-phase route towards hydroxylamines via an oxidation–Cope elimination began by attaching acryloyl chloride to hydroxymethyl polystyrene in order to produce the REM resin. The Michael addition of a secondary amine produced the corresponding tertiary β-amino ester. After washing the resin, this tertiary amine was reacted with mCPBA in chloroform for 1–2 h to form the N-oxide, which in turn underwent an oxidation–Cope elimination to produce the N,N-disubstituted hydroxylamine and regenerate the acrylate resin. Sammelson and Kurth34 also examined other oxidation protocols viz., H2O2 in THF, peracetic acid in various solvents, and dimethyldioxirane in acetone/CH2Cl2 and came to the conclusion that these procedures were not as effective as mCPBA/CHCl3.
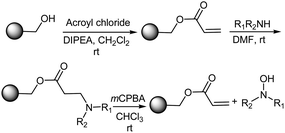 |
| | Scheme 6 The solid-phase synthesis of hydroxylamines via an oxidation–Cope elimination. | |
2.4. Rubottom oxidation
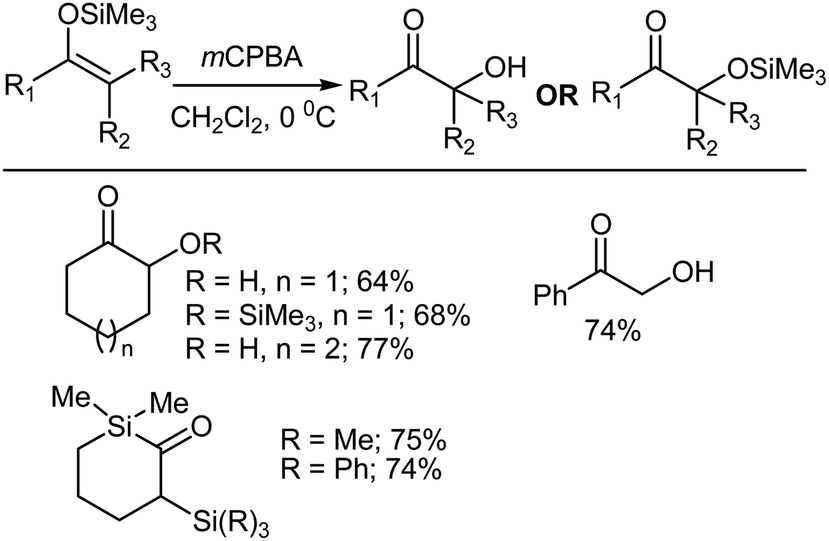
The synthesis of α-hydroxy ketones can be achieved by reacting silyl enol ethers with mCPBA, followed by a subsequent rearrangement. Aqueous work up gives the desired product after desilylation.35–38 Silyl enol ethers are readily prepared from enolizable ketones using a base and chlorosilane.39–41 The silyl enol ethers are usually treated with a slight excess of mCPBA in CH2Cl2 at 0 °C followed by work up involving the addition of pentane. Perusal of Scheme 7, which summarizes several representative Rubottom oxidation products, reveals that the introduction of the hydroxy group is regiospecific and that no exchange occurs with respect to the position of the original carbony1 group in the ketonic precursor. However, a non-aqueous work up of the oxidation products yielded α-trimethylsiloxy ketones.35
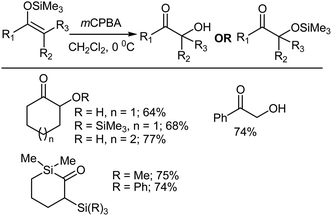 |
| | Scheme 7 Rubottom oxidation using mCPBA. | |
2.5. Nef reaction
The conversion of a nitro group into a carbonyl group has been firmly established and applied since its discovery by Nef more than a century ago, and is regarded as one of the most important functional group transformations in synthetic organic chemistry. The success of this procedure has been verified by the large number of different synthetic protocols that have been devised over the years in order to accomplish this transformation with an ever increasing level of chemoselectivity.42 The Nef reaction often involves either strongly acidic or strongly basic conditions,43–48 but it is well known that trialkylsilyl enol ethers (which can be generated from trialkyl chloride and DBU) readily undergo α-hydroxyalkylation by means of mCPBA under extremely mild conditions.35,37,38,49 Therefore, Aizpurua et al.50 reacted different nitroalkanes with trialkyl chloride and DBU, followed by the oxidative cleavage of the resulting trialkylsilyl enol ethers with mCPBA to give ketones (method A).50,51 In another study, Kim and Oh52 reported the conversion of nitro compounds to carbonyl compounds (the Nef reaction) using TiCl4 and mCPBA (method B). Various structurally diverse secondary nitro compounds have thus been converted to carbonyl compounds using both methods A and B (Scheme 8).
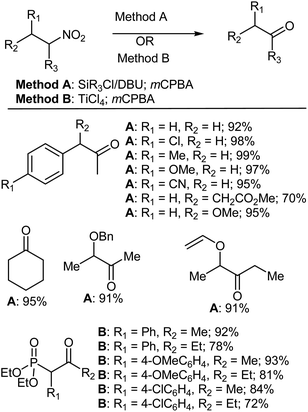 |
| | Scheme 8 The Nef reaction with mCPBA. | |
2.6. Hofmann rearrangement
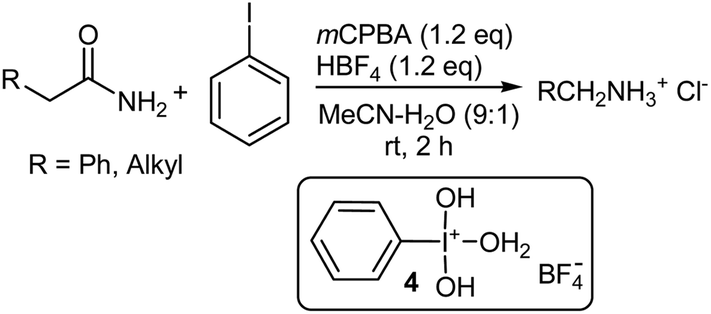
The classical Hofmann rearrangement involving sodium hypobromite or hypochlorite under basic conditions affects the conversion of primary carboxamides to primary amines that possess one less carbon atom, through the intermediacy of either an N-bromo- or N-chloroamide.53–55 Tetra-coordinated bis(aqua)(hydroxy)phenyl-λ3-iodane complex 4,56,57 generated from the reaction of a stoichiometric amount of iodobenzene with mCPBA in aqueous acetonitrile in the presence of 48% aqueous HBF4, was found to initiate the Hofmann rearrangement of α-phenylacetamide (Scheme 9).58 The reaction took place smoothly at room temperature and was finished within 2 h. Subsequent treatment with an aqueous HCl solution, afforded the rearranged benzylammonium chloride quantitatively. The introduction of both electron-donating (p-Me, 3,5-Me2 and 2,4,6-Me3) and electron-withdrawing groups (p-Cl and p-CF3) into the iodobenzene ring decreased the yield. Aliphatic iodides such as methyl, trifluoroethyl, and 1-adamantyl iodide exhibited no catalytic efficiency. This new catalytic method has found general use in the Hofmann rearrangement of primary carboxamides and all of the unfunctionalized simple linear, branched, and cyclic aliphatic carboxamides examined afforded the corresponding alkylammonium chlorides with one less carbon atom at room temperature and in high yield. The catalytic conditions are compatible with the presence of various types of functionality including halogens (F, Cl, Br), sulfonamides, amines, methoxy and nitro groups.58
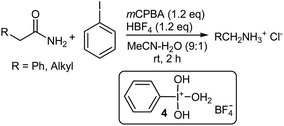 |
| | Scheme 9 The Hofmann rearrangement of α-phenacylacetamides with mCPBA. | |
2.7. Dakin oxidation
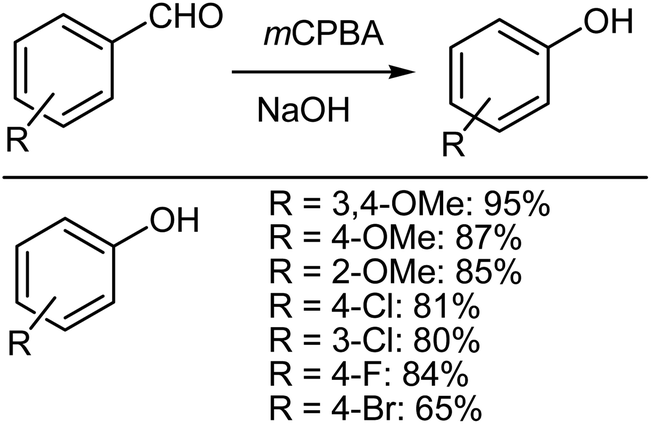
The Dakin reaction formally involves the conversion of hydroxybenzaldehydes into phenols using alkaline hydrogen peroxide.59 Phenols are suitable intermediates for many purposes and have proven to be useful in the synthesis of several biologically active compounds.60 Although there are several different methodologies focused on the preparation of phenols from benzaldehydes,61–67 Fraga et al.68 reported on a mCPBA mediated Dakin reaction that had the advantage of requiring shorter reaction times and an easier work up. Fraga et al.68 described a solid-state Dakin oxidation using mCPBA and various non-activated benzaldehydes, which could all be converted into their corresponding phenols with a remarkable reduction in the reaction time and in high yield, compared with other previously reported standard type methodologies.61–67 The simple, but very careful mixing of an aromatic aldehyde and mCPBA using a pestle and mortar results in a pasty mass that, after a few minutes, yields the desired phenol derivative, which can be easily isolated after a sequence of alkaline hydrolysis (Scheme 10).69,70
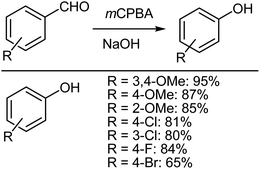 |
| | Scheme 10 Dakin oxidation of aromatic aldehydes using mCPBA. | |
2.8. Kita lactonization
A variety of natural products bearing spirocyclic systems exist, and many of them are biosynthetically formed via an oxidative spiroannulation processes.6,71,72 Hypervalent iodine(III) reagents are generally considered to be one of the most effective oxidants to affect these oxidation processes. In evaluating their low toxicity, the method has been widely used for the total synthesis of natural products having important biological properties such as antitumor, antibacterial, antifungal, and antiprotozoan activities.73–75 mCPBA is one of only a few chemical oxidants that can convert iodine compounds to their corresponding iodine(III) forms selectively in organic solvents at room temperature.76–83 Kita and co-workers reported on the enantioselective oxidative dearomatization of 1-naphthol derivatives to spirolactones with high enantioselectivities (up to 69% ee) using stoichiometric amounts of a chiral iodine(III) reagent 6, which was generated in situ from 5 and mCPBA in the presence of acetic acid (Scheme 11).84,85 In another study, Ishihara et al.86 reported the conformationally flexible C2-symmetric chiral iodoarene 7 as a highly effective precatalyst that, in the presence of mCPBA, acts as a terminal oxidant for enantioselective Kita oxidative spirolactonization (Scheme 11).86 In their report, they described a detailed investigation into enantioselective oxidative spirolactonization. A broad range of substrates and products have been described with high enantioselectivities of up to 92% ee using 7 and mCPBA.87
 |
| | Scheme 11 Enantioselective Kita oxidative spirolactonization of 1-naphthols. | |
3. Halogenation
3.1. Halogenation of pyrimidine and purine derivatives
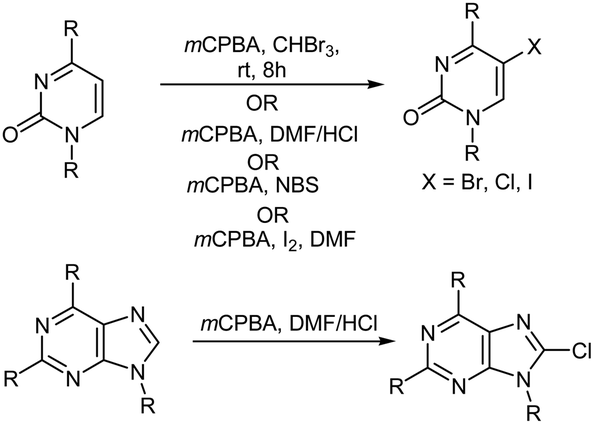
The selective oxidative halogenation of organic compounds in a simplified manner has received considerable interest in recent years.88 Numerous methods have been reported89–95 for the halogenation of uracil because it is a fundamental building block for nucleic acids96 and halogenated uracils are important due to their use in medicinal chemistry.97 Molte-Leth and Jorgenson88 reported on the oxidative bromination of uracil using mCPBA and CHBr3. In another study, Ryu and MacCoss98 reported on the synthesis of 5-chloro-substituted pyrimidine nucleosides and 8-chloro-substituted purine nucleosides using mCPBA in an aprotic solvent such as dimethylformamide (DMF). The chlorination of different pyrimidine and purine derivatives was also achieved after reaction with mCPBA in dipolar aprotic solvents containing HCl. In the pyrimidine series, both uracil and cytosine derivatives gave the corresponding 5-chloro derivatives in high yields. Application of the same reaction conditions to the purine derivatives adenosine and guanosine, afforded the 8-chloro nucleosides in good yield.98 Similarly, Zanatta et al.99 reported the preparation of 5-bromo-substituted pyrimidine nucleosides by using mCPBA and NBS while Ryu et al.100 reported the preparation of 5-iodo-substituted pyrimidine nucleosides by using mCPBA, I2, and DMF (Scheme 12).
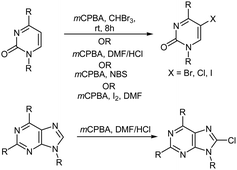 |
| | Scheme 12 Halogenation of pyrimidine and purine derivatives. | |
3.2. Halogenation of organic compounds with a mCPBA–HCl–DMF system
3.2.1. Halogenation of aromatic compounds. Since the middle of 19th century, the chlorination of phenols has been extensively described using an extensive array of different reagents.101–111 Ryu et al.112 reported the chlorination of phenols under mild conditions using a mCPBA–HCl–DMF system (Scheme 13). Reaction of 2-naphthol, 2-chlorophenol, 4-chlorophenol with an equimolar amount of HC1 and mCPBA in DMF gives the corresponding monochloro-substituted phenols in good yields with high regioselectivity. The monochlorination of phenol ethers, such as anisole, also proved to be equally effective. Accordingly, the monochlorination of chlorophenols, such as 2- and 4-chlorophenols, afforded excellent yields of the dichloro-substituted compounds. Dichloro or trichloro-compounds were also obtained by the reaction of phenol, o-cresol, 1-naphthol as well as ethyl salicylate with a two or three-fold excess of HC1 and mCPBA, in high yields and with high regioselectivity.112
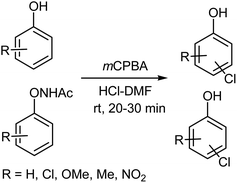 |
| | Scheme 13 Chlorination of aromatic compounds with the mCPBA–HCl–DMF system. | |
Despite there being numerous methods for the chlorination of acetanilides,113–119 Ryu et al.120 used the mCPBA–HCl–DMF system under mild conditions (Scheme 13) to effect the monochlorination of various acetanilides, such as acetanilide, 2-methylacetanilide, 3,5-dimethylacetanilide, and 2-chloroacetanilide, which gave the corresponding monochloroacetanilides in good yields with high regioselectivity along with the dichloroacetanilides. In the case of 4-methylacetanilide and the nitroacetanilides, the corresponding monochloroacetanilides were only obtained in moderate yields. It is noteworthy that in the case of 2,4-disubstituted acetanilides, the desired chloro compounds were obtained in poor yields. Dichlorination of acetanilides was also affected using a two-fold excess of HC1 and mCPBA, producing the corresponding dichlorinated compounds in good yields and high regioselectivity.120
3.2.2. α-Halogenations. A wide variety of synthetic reagents and methods are currently available for the synthesis of α-chloroketones121 and the large majority of these chlorination methods involve the α-chlorination of ketones, whereas only a few examples of the direct conversion of secondary alcohols into α-chloroketones have been reported.122–125 Ryu et al.126 found that secondary benzylic alcohols were easily oxidized concomitantly in situ using the mCPBA–HCl–DMF system to yield α-chloroketones in good yields (Scheme 14). It was found that the reaction initially involved the rapid transformation of the alcohol into a ketone followed by α-chlorination. In cases where electron-donating groups (hydroxy and alkoxy groups) were present, a mixture of several compounds was obtained due to chlorination of the aromatic ring. It is interesting to note that the molar ratio of mCPBA to HC1 was (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.3) and by adding one additional equivalent of mCPBA, higher yields and minimized amounts of the side products could be obtained.126
:3.3) and by adding one additional equivalent of mCPBA, higher yields and minimized amounts of the side products could be obtained.126
 |
| | Scheme 14 α-Chloroketones from secondary benzylic alcohols using the mCPBA–HCl–DMF system. | |
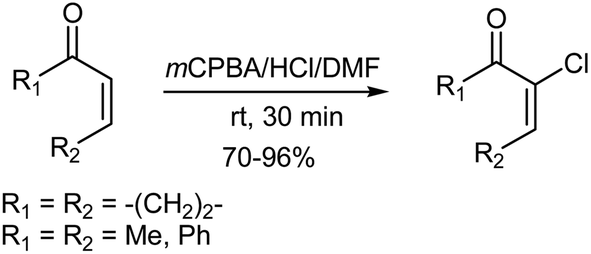
In another study, Ryu et al.127 used the mCPBA–HCl–DMF system for the chlorination of α,β-unsaturated enones, synthesising α-chloro-α,β-unsaturated enones in good yields and with high regioselectivity (Scheme 15). Different oxidizing agents such as Oxone and ammonium cerium nitrate were also tried, but these reagents gave unsatisfactory results.
 |
| | Scheme 15 Chlorination of α,β-unsaturated enones with mCPBA–HCl–DMF. | |
Similarly, Ryu et al.128 used mCPBA–HCl–DMF (method A) for the chlorination of ketones, while Inukai et al.129 used mCPBA/MgBr2 (method B) for the corresponding bromination of ketones. It is interesting to note that when either the reaction temperature or the molar ratio of HCl–DMF to the substrate were raised in method A, the formation of side products increased and, subsequently, the yield of the α-haloketones was decreased (Scheme 16).
 |
| | Scheme 16 The α-halogenation of ketones. | |
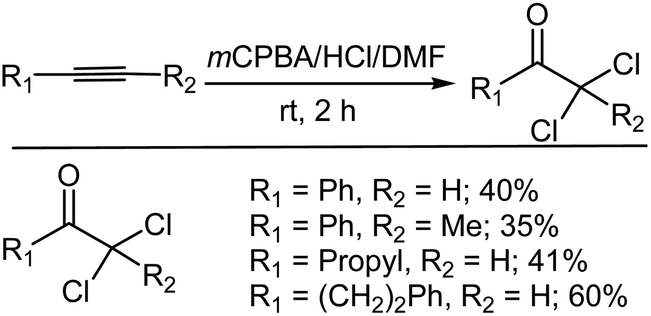
3.2.3. Chlorination of alkynes. Ryu et al.130 also reported the reaction of various alkynes with mCPBA–HCl–DMF, which provided the corresponding α,α-dichloroketones in moderate yields (Scheme 17). Ryu et al.130 also attempted the same reaction using other oxidants such as hydrogen peroxide and Oxone. The group found that Oxone gave the best results. However, by using mCPBA the formation of α-monochloroketones was increased.
 |
| | Scheme 17 Oxidative chlorination of alkynes with mCPBA–HCl–DMF. | |
3.2. Miscellaneous halogenations
Treatment of p-alkylbenzenesulfonic acids with mCPBA and molecular iodine gave p-alkyliodobenzenes in good to moderate yields via an electrophilic ipso-substitution of the iodonium species (I+) formed (Scheme 18). Interestingly, this desulfonyloxyiodination was promoted by the addition of a catalytic amount of an iodoarene, such as o-iodobenzoic acid.131 Similar treatment of dimethylbenzenesulfonic acids and trimethylbenzenesulfonic acids with mCPBA and molecular iodine proceeded smoothly, both in the absence and presence of o-iodobenzoic acid, providing the corresponding monoiodo-dimethylbenzene and diiodo-dimethylbenzene, and monoiodo-trimethylbenzene and diiodo-trimethylbenzene, in good to moderate yields, respectively. Conversely, the same desulfonyloxyiodination of benzenesulfonic acid and p-chlorobenzenesulfonic acid with mCPBA and molecular iodine proceeded only in the presence of o-iodobenzoic acid, generating iodobenzene and p-chloroiodobenzene, respectively, in moderate yields.131
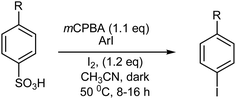 |
| | Scheme 18 Desulfonyloxyiodination and iodination with mCPBA/ArI/I2. | |
In another study, it was reported that the aryl ring of several types of phenyl ethers could be monobrominated in good yields with potassium bromide in the presence of 18-crown-6 during oxidation with mCPBA (Scheme 19).107 Similarly, monoiodination was also possible with both phenyl ethers and free phenols by using potassium iodide in the presence of 18-crown-6 during oxidation with mCPBA.
 |
| | Scheme 19 The halogenation of phenyl ethers. | |
4. Olefin functionalization
4.1. Epoxidation of olefins
Despite the development of many new oxidation procedures, the use of peroxy acids such as mCPBA still constitutes one of the most useful synthetic procedures for the epoxidation of alkenes on a laboratory scale132,133 Oxygen atom transfer from mCPBA to an alkene is facilitated by electron-donating substituents on the carbon–carbon double bond and electron-withdrawing groups on the peroxy acid.134–143 High yields have been obtained for a variety of substituted olefins (Fig. 1)144–198 and there are numerous reports199–305 in which mCPBA is used for epoxidations of different substrates.
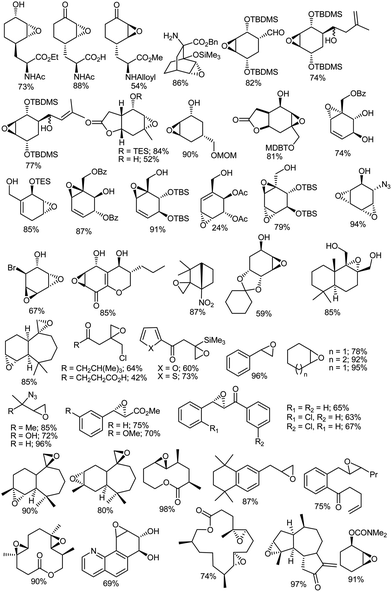 |
| | Fig. 1 mCPBA epoxidation products of different alkene substrates. | |
4.2. 1,2-Dioxygenation of olefins
4.2.1. Dihydroxylation. Perhydroxylation of carbon–carbon double bonds is a process of most vital synthetic and theoretical interest and, therefore, many reagents and procedures have been investigated in the hope of obtaining highly stereoselective reactions with high yields.306 The anti 1,2-dihydroxylation process can be achieved using a vast number of different methods307–312 but sometimes these methods give mixtures of the diol and its corresponding esters and thus further hydrolysis is necessary to obtain the pure diol. This additional inconvenience reduces the overall yield and increases the time of the reaction. To overcome these problems, Fringuelli et al.313 reported a very simple procedure, which allows one to prepare anti 1,2-diols in excellent yields in a one-pot-two step synthesis that does not require organic solvents (Scheme 20). In this protocol the alkene is first epoxidized by mCPBA in deionized water, which is then followed by ring opening of the epoxide by either acid or basic hydrolysis. High yields were always observed (80–95%) and the diol was the only reaction product isolated (Scheme 20). The epoxidation stage occurs in water under very mild conditions and, therefore, this one-pot process is suitable for acid sensitive alkenes.313 In another study, Davies et al.314 reported the 1,2-dihydroxylation of N,N-dibenzylcyclohex-2-enamine using Cl3CCO2H and mCPBA.
 |
| | Scheme 20 Synthesis of anti 1,2-diols in water via epoxidation–hydration reactions. | |
Mayer et al.315 also reported the cis-dihydroxylation of olefins using a Tp–osmium complex (8), mCPBA, and HCl (Scheme 21). Different alkenes were subjected to dihydroxylation under the same conditions and, interestingly, for all of the alkenes, dihydroxylation occurs in a cis fashion. Styrene, trans-4-dimethylamino-4′-nitrostilbene, cyclohexene, trans-dimethyl fumarate and trans-methyl cinnamate were all converted to their respective cis-diols. It is interesting to note that the reaction proceeds well with hydrocarbons, electron-deficient alkenes, and an alkene with an electron-donating dimethylaniline substituent.
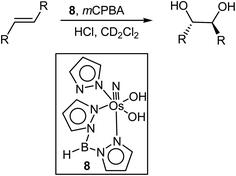 |
| | Scheme 21 cis-Dihydroxylation of olefins with 8 and mCPBA. | |
4.2.2. Diacetoxylation and lactonization of olefins. Gade et al.316 reported the clean and efficient trans-diacetoxylation reaction of alkenes catalyzed by triflic acid using mCPBA as the oxidant (method A, Scheme 22). Under these conditions, both linear and cyclic aliphatic alkenes were converted to their corresponding diacetoxylation products in good to excellent isolated yields. In general, terminal alkenes and cyclic alkenes also gave their corresponding diacetoxylation products in good to high yields. Conversely, when the double bond was not terminal, a relative decrease in yield was observed and the same was also found for aromatic alkenes such as styrenes. Recently, a novel method for the organocatalytic syn diacetoxylation of alkenes has been developed by Li et al.317 using 4-MeC6H4I as an efficient catalyst and mCPBA as an oxidant (method B, Scheme 22). A broad range of substrates, including electron-rich and electron-deficient alkenes, were smoothly transformed, furnishing the desired products in good to excellent yields with high diastereoselectivity (up to >19:1 dr).
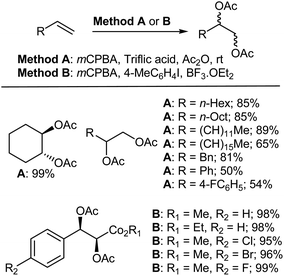 |
| | Scheme 22 Diacetoxylation of alkenes using mCPBA as oxidant. | |
Method A was also very efficient for the oxidative lactonization of ω-unsaturated carboxylic acids to afford five-membered lactones (Scheme 23). Notably, 4-phenylpentenoic acid gave the corresponding lactone in moderate yield, whereas only the rearrangement product due to 1,2-phenyl migration was isolated in the TfOH-catalyzed lactonization using PhI(OAc)2 as the oxidant.318 5-Hexenoic acid and 6-heptenoic acid failed to afford the corresponding lactones, but yielded the linear diacetoxylation products instead.
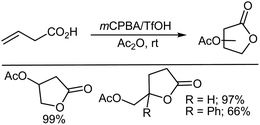 |
| | Scheme 23 Lactonization of alkenes using mCPBA as oxidant. | |
4.3. Aziridination of olefins
The aziridination of alkenes is an important chemical transformation and is a convenient method for accessing various biologically active nitrogen-containing products and synthetic intermediates.319–322 Zhdankin et al.323 reported a metal-free catalytic aziridination using a catalytic amount of tetrabutylammonium iodide (TBAI), mCPBA as a terminal oxidant, and PhthNH2 as a nitrenium precursor (Scheme 24). Various substituted alkenes were transformed into their respective aziridines in good to excellent yields. In general, all of the styrenes investigated with either electron-donating or electron-withdrawing substituents afforded products in good yields. This reaction also gave good yields with α- or β-substituted styrenes. In reactions involving non-hindered cycloalkenes, the products were obtained in moderate yields, although a substituted cycloalkene (1-methylcyclohexene) and seven- or eight-membered cycloalkenes gave lower yields of their respective aziridines. The reactions of 1-decene and α,β-unsaturated cyclic ketones under these conditions afforded the corresponding aziridines in low yield. Unfortunately, these yields were not improved upon using a stoichiometric amount of TBAI.
 |
| | Scheme 24 Aziridination of alkenes. | |
5. Oxidation of functional groups
5.1. Oxidation of alcohols
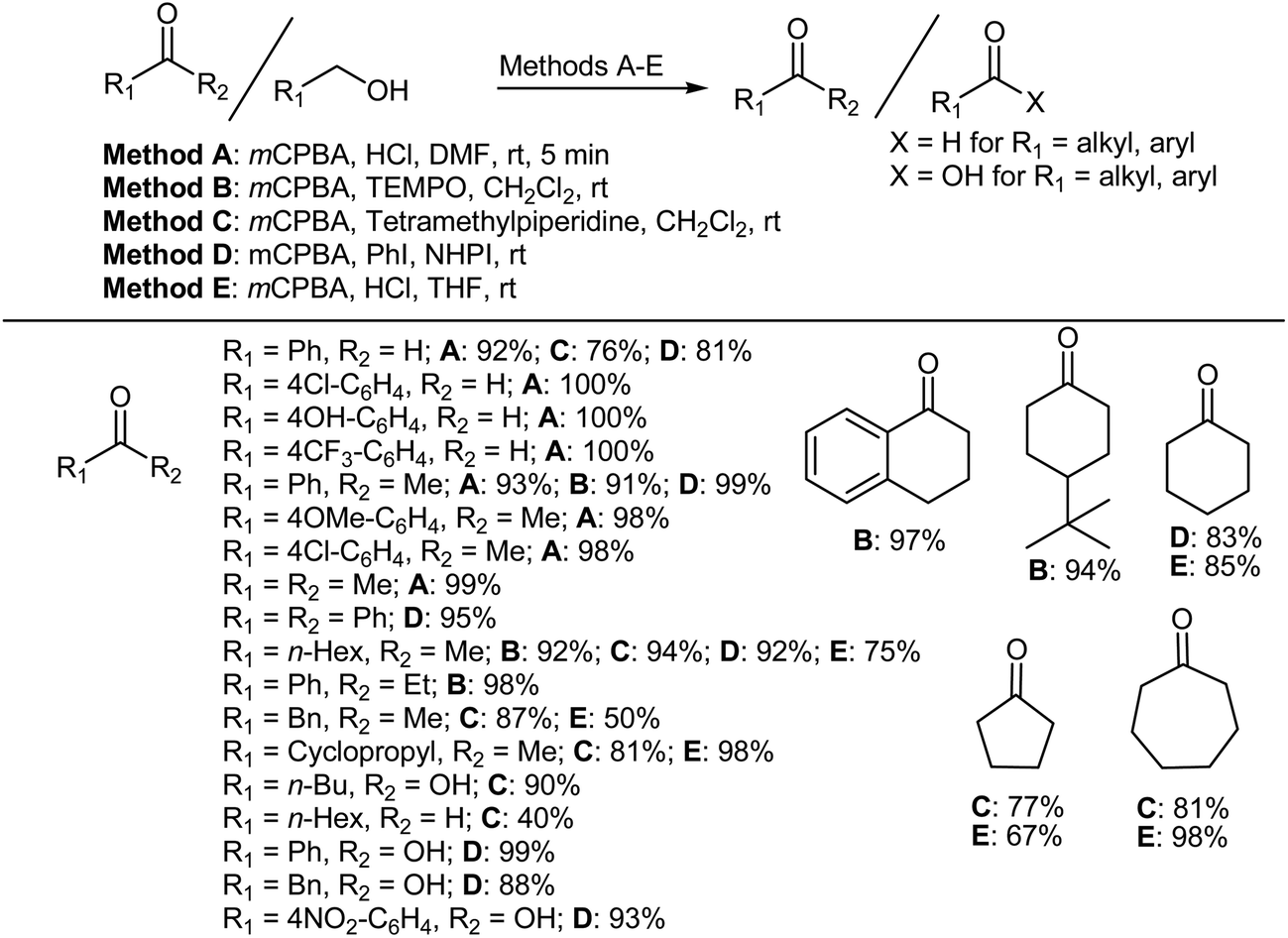
The oxidation of alcohols is probably one of the most important protocols in organic synthesis and since carbonyl compounds are widely used as intermediates both in manufacturing and the laboratory,324 the development of new oxidative protocols continues to receive renewed attention, in spite of the availability of several methods to achieve such objectives. Interestingly, method A (mCPBA, HCl, and DMF),325 method B (mCPBA, TEMPO, CH2Cl2),326 method C (mCPBA, tetramethylpiperidine, CH2Cl2),327,328 method D (mCPBA, PhI, N-hydroxyphthalimide [NHPI]),329 and method E (mCPBA, HCl, THF)330 shown in Scheme 25 can be used to selectively oxidize primary alcohols to their corresponding aldehydes and carboxylic acids and secondary alcohols (benzylic, aliphatic, alicyclic, and heterocyclic alcohols) to their corresponding ketones in excellent yields. It is noteworthy that, with the exception of the case of cyclohexanol, no Baeyer–Villiger oxidation of the ketonic products was encountered using the conditions in method C. This is not surprising, since the Baeyer–Villiger oxidation generally requires longer reaction times or higher temperatures and employs stronger peracids than those needed for alcohol oxidation. Cyclohexanol (a notable exception) is considerably more reactive than its cyclic congeners towards the Baeyer–Villiger oxidation. For the resultant cyclohexanone, it was possible to suppress or enhance the Baeyer–Villiger oxidation by proper choice of the reaction conditions. In general, the Baeyer–Villiger oxidation of the ketonic products can be avoided by conducting the reaction under mild conditions. Method D was particularly effective for the chemoselective oxidation of primary alcohol functionality in the presence of secondary alcohol functionality. In methods C and D the primary alcohols were also oxidized to carboxylic acids. A limitation of method E is that the reaction conditions are unsuitable for the oxidation of acid sensitive compounds.
 |
| | Scheme 25 Selective oxidation of primary and secondary alcohols to their corresponding aldehydes (or carboxylic acids) and ketones. | |
5.2. Oxidation of ethers
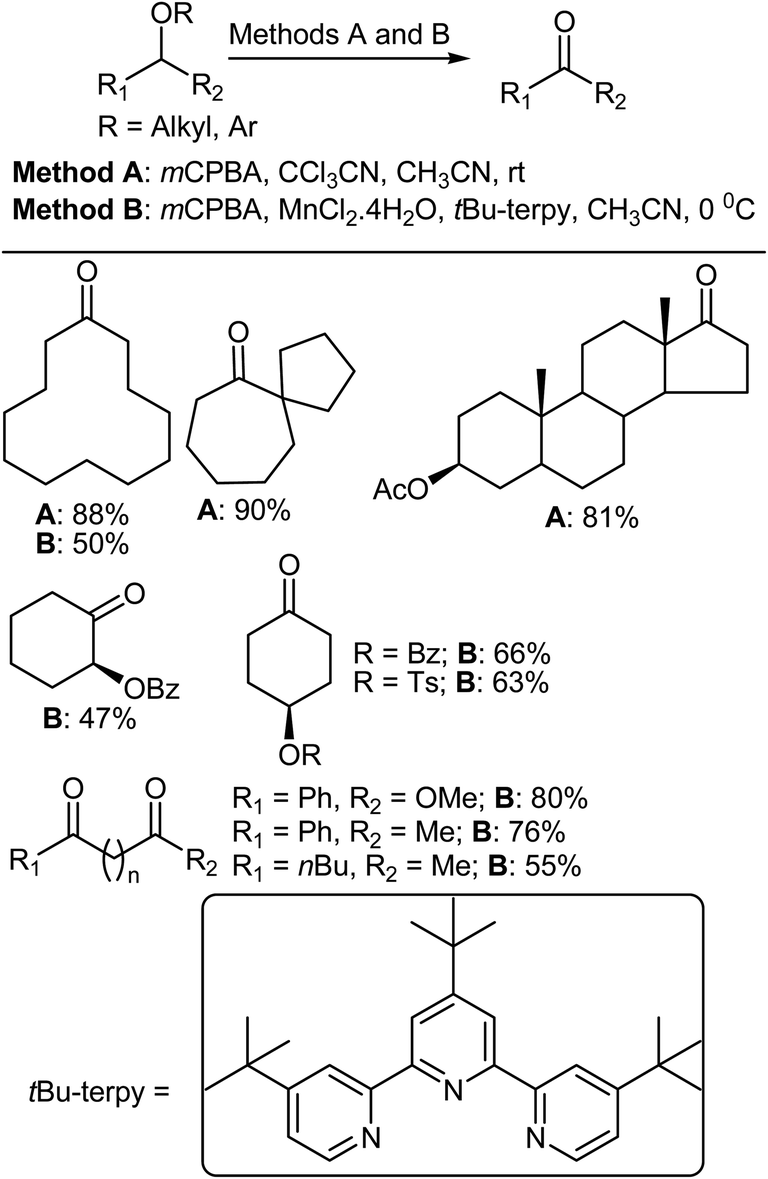
The oxidation of organic compounds is of general fundamental importance in chemistry.331 Inoue et al. reported two methods for the direct oxidation of ethers to ketones. Method A (mCPBA, CCl3CN, MeCN)8 and method B (mCPBA, MnCl2·4H2O, t-Bu-terpy, CH3CN)332 could be used to chemoselectively convert alkyl ethers to ketones under mild conditions (Scheme 26). In order to explore the substrate scope and chemoselectivity of method A, various differently substituted ethers were treated using optimized reaction conditions. Initially, the oxidation of cyclododecyl ethers was investigated and, similar to the case of methyl ether, oxidations of the octyl, isopropyl, and benzyl ethers all produced ketones. Meanwhile, the sterically more demanding t-butyl group impeded the oxidation. However, 4-pentenyl ether and its cyclohexanone analogue were both converted into their respective ketones. Similarly, methyl ether substituted carboskeletons were subjected to the same reaction conditions and oxidation of the seven-membered methoxy ring gave the ketone in high yield. Importantly, only the methyl ethers of differentially protected cyclohexane diols were oxidized to carbonyl groups. When electron-withdrawing acetyl and mesyl groups and sterically bulky TBDPS and trityl groups were used as ether groups the hydroxyl functionality was effectively protected, demonstrating the high chemoselectivity of the reaction.
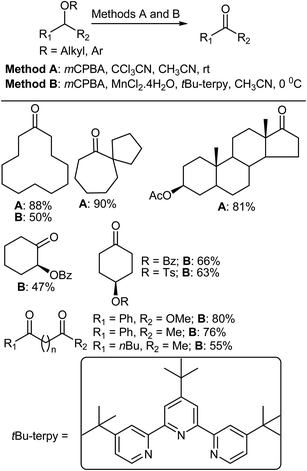 |
| | Scheme 26 The oxidation of various ethers. | |
Various cyclododecanol derivatives were also prepared and subjected to oxidation using method B. It was found that the oxidation of cyclododecyl octyl ether proceeded in a similar manner to that of the methyl ether, and formation of the ketone and octanoic acid was confirmed by analysis of the crude reaction mixture. The observation octanoic acid formation indicated that octan-1-ol that was eliminated from the cyclododecanol and further oxidized to give octanoic acid under the reaction conditions. The oxidation of benzyl ethers gave the respective ketones as the major product in 46–55% yield. In addition, the oxidation of electron-rich 4-methoxybenzyl ether was completed in a shorter reaction time (1 h at 0 °C) compared to the other benzyl ethers, although a similar yield of ketone was obtained irrespective of the electronic properties of the aromatic ring.
5.3. Oxidation of amines
5.3.1. Oxidation of amines to nitro compounds. Nitroalkanes are a versatile class of materials that can be used as intermediates in chemical transformations (e.g., the Nef and Henry reactions). They are also frequently used for the preparation of pharmaceuticals, dyes and agrochemicals. The nitro derivatives of aliphatic caged compounds, such as cubane or adamantane, have been explored as highly energetic materials.333 Although the synthesis of primary and secondary nitroalkanes from amines using mCPBA and 1,2-dichloroethane (DCE) has been reported by Gilbert and Borden,334 Schreiner et al.333 found that this procedure was also applicable for the preparation of nitro diamondoids (Scheme 27). During their synthesis of unequally disubstituted diamantane derivatives Schreiner et al. utilized mCPBA to oxidize the amino group, preparing the corresponding nitro compounds. A diamondoid amine can be fairly easily be prepared either by chloroacetamidation of the alcohol followed by acidic cleavage using thiourea or by treatment of the carboxylic acid with diphenylphosphoryl azide followed by hydrolysis.335 This has thus made it possible for various diamondoid nitro derivatives to be synthesized using the mCPBA oxidation of the corresponding amines.
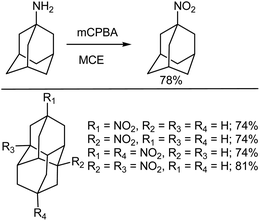 |
| | Scheme 27 Synthesis of diamondoid nitro compounds from amines with mCPBA. | |
5.3.2. Oxidation of amines to nitroso compounds. From the late 1960s onward, there have been many reports of the use of mCPBA for the synthesis of nitroso compounds (Scheme 28). A particular example of such a transformation is the oxidation of 3-aminobenzamide using mCPBA in DMF at 0–5 °C to give 3-nitrosobenzamide.336,337 Preparations of 6-nitroso-1,2-benzopyrone, 5-nitroso-1(2H)-isoquinolinone, 7-nitroso-1(2H)-isoquinolinone, and 8-nitroso-1(2H)-isoquinolinone have also been reported. These aryl nitroso compounds were prepared in order to test their suitability as specific inactivators of retroviral zinc fingers and as antitumor agents.336 The preparation of 2,6-dimethyl-, 2,6-diethyl- and 2-ethyl-6-methylnitrosobenzene using dichloromethane as solvent has also been reported338 and this reaction has been extended to use acetonitrile-d3 as a solvent to prepare the same products and 2-methyl-6-tert-butylnitrosobenzene. It was also found that very low yields of <5% of the corresponding nitroso products were obtained upon oxidation of the 2-chloro-4-methylaniline and 2,4-dimethylaniline precursors.339
 |
| | Scheme 28 Conversion of amines to nitroso compounds with mCPBA. | |
The reaction of mCPBA with aliphatic primary amines viz., 2-butylamine, 1-hexylamine, 1-propylamine, 2-phenylethylamine and cyclohexylamine in dichloromethane at room temperature has been shown to give excellent yields of the dimeric nitroso compounds.334 Baer and Chiu340 prepared a number of dimeric nitroso sugars by adding a chloroform or chloroform–methanol solution of the amino sugar dropwise to a refluxing solution of mCPBA in chloroform. Furthermore, trans-dimeric 2-nitrosocyclohexanol could also be prepared using the same method. Several C-nitroso compounds such as nitrosomesitylene, 2-nitrosotoluene, nitrosocyclohexane, 1-alkyl-1-nitrosocyclohexane (where alkyl = Me, Et or cyclohexyl), 1-ethyl-1-nitrosocyclopentane, nitroso-tert-butane, 2-nitrosoisocamphane, and 2,2,4-trimethyl-4-nitrosopentane have been prepared using mCPBA as the oxidant.337,341
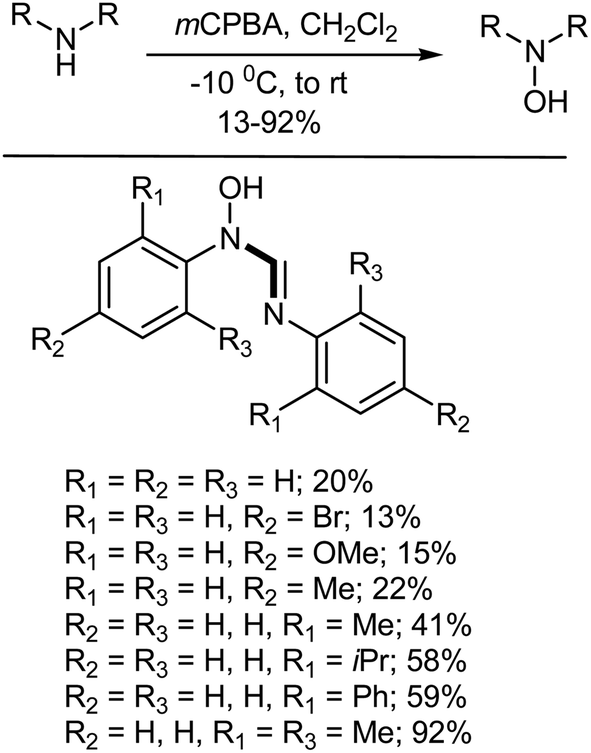
5.3.3. Oxidation of amines to hydroxylamines. N,N′-Disubstituted hydroxyamidines/amidoximes have been studied for their biological activity (as antitubercular agents and hypotensives) and pharmacological properties (such as their bactericidal, fungicidal and local anaesthetic properties, to mention just a few).342 They have also found good use as precursors in the synthesis of cyclic compounds.343,344 The N-oxidation of N,N′-disubstituted amidines with mCPBA provides a mild, rapid, and efficient route to the corresponding hydroxyamidines (Scheme 29). The efficiency of the N-oxidation reaction is influenced by the substitution on the N,N′-aryl rings. Substrates with electron-donating bis-ortho substituents give, in general, good yields (88–92%), while only moderate yields (41–59%) were obtained for compounds bearing electron-donating mono-ortho substituents. An absence of ortho substitution results in rather poor yields (13–22%) as the compounds decompose during purification. The effect of ortho substitution on the efficiency of the reaction could be explained by the higher stability of the o-substituted compounds, in which the central carbon is best protected on steric grounds. Electron-donating ortho substituents also increase the basicity of the imido-nitrogen, which has plays the determining role in driving the N-oxidation reaction due according to electronic considerations. For very bulky ortho-substituted substrates, steric hindrance starts to reduce in the amount of product formed in the N-oxidation reaction as well.342
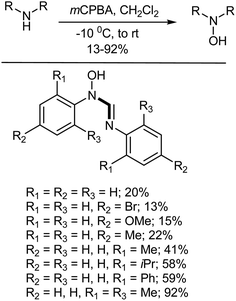 |
| | Scheme 29 Synthesis of hydroxyformamidines via N-oxidation of the corresponding formamidines. | |
6. Unnamed rearrangements
6.1. Oxidative rearrangement of ketimines to amides
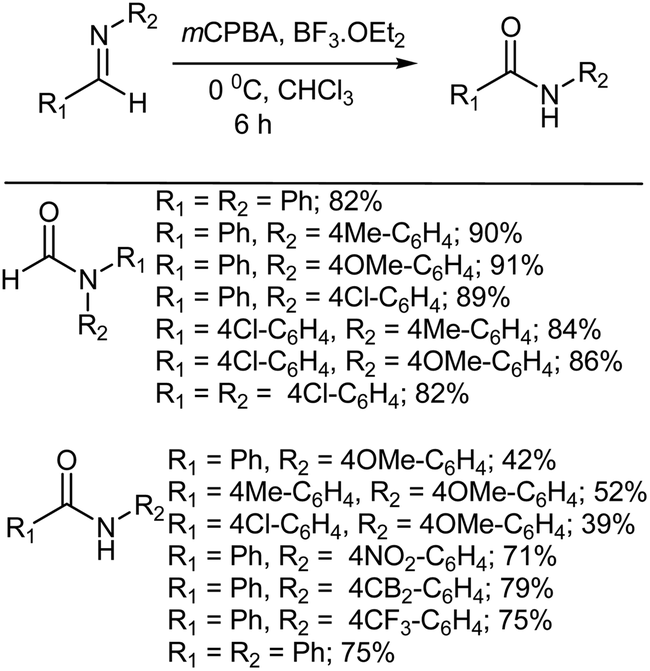
The amide moiety is one of the most popular and important functional groups and is used by many research groups. The amide unit is frequently found in a variety bioactive natural compounds and it can be a valuable intermediate for the preparation of many other functional groups, such as amines, aldehydes and acids, to name but a few.345 Conversion of ketones to amides has been extensively reported using a wide range of different methods and reagents.346–372 Rhee and coworkers373 reported an oxidative rearrangement of alkyl aryl ketimines to amides using mCPBA and BF3·OEt2 (Scheme 30). It was found that, during the reaction, an aryl group migrated from carbon to the nitrogen atom every time and that the product formed from the reaction was strongly influenced by the difference in migratory aptitude between the aryl and alkyl groups, conforming to the results of the Baeyer–Villiger oxidation. The alkyl group migration of ketimines was not observed at all. Interestingly, in the presence of p-toluidine the yield of the oxidative rearrangement dropped due to greater decomposition of the ketimine with mCPBA. The electronic effect of a para substituent on the migrating aryl group of the ketimine itself did not affect the reaction yield all that much. However, the presence of an electron withdrawing substituent on the aryl ring was inclined to lower the yield of the product compared to H and electron-donating groups.
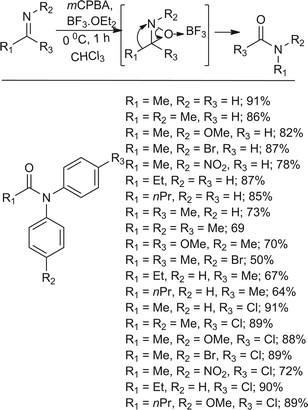 |
| | Scheme 30 Oxidative rearrangement of ketimines to amides with mCPBA and BF3·OEt2. | |
6.2. Oxidative rearrangement of aldimines to amides
Rhee and coworkers also reported the oxidative rearrangement of N-benzylaldimines to N-benzylamides using a combination of mCPBA and BF3·OEt2 (Scheme 31). N-Benzylamide was obtained in every reaction along with some recovered arylaldehyde.374 The electronic effects of substituents on the aryl group of the benzaldimines did not affect the reaction yields to any great extent.374
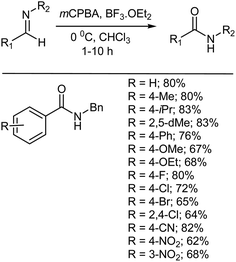 |
| | Scheme 31 Oxidation of N-benzylaldimines to N-benzylamides by mCPBA and BF3·OEt2. | |
In another study, Rhee et al.375 reported the oxidation of a different group of aldimines to afford amides by using a similar combination of mCPBA and BF3·OEt2 (Scheme 32). This study indicated that the reaction product was strongly influenced by the electron releasing capacity of the aromatic substituent. In the case of electron-releasing substituents on the aryl group, the oxidation of imines afforded formamides, in which aryl group migration had occurred. However, in the case of an electron-withdrawing substituent being on the aryl group the reaction provided the amide, but formed via an alternative hydride migration. In the case of a chloro substituent, the formamide was obtained as the major product (83% yield) along with 5.6% of the amide. Some researchers have suggested376–378 that oxaziridine ring formation may be involved in the reaction, but the actual mechanism has not been determined.
 |
| | Scheme 32 Oxidation of aldimines to amides by mCPBA and BF3·OEt2. | |
In contrast to this, when p-anisidine was used as the aldimine, the oxidation affords only the N-(p-methoxyphenyl)-p-substituted-benzamide along with a considerable amount of recovered arylaldehyde. It was presumed that the reaction follows an internal hydrogen abstraction and undergoes decomposition to the corresponding aldehyde. The electron releasing group, i.e. the methoxy group of p-anisidine, increases the electron density on the nitrogen atom and aides the coordination of the Lewis acid to the lone pair of the nitrogen atom. After the formation of the peroxy intermediate by attack of mCPBA on the iminium carbon, rapid fragmentation of the peroxy intermediate presumably occurs to provide the amide.375
6.3. Oxidative rearrangement of cyclic enol ethers to α-alkoxyesters
Wipf and coworkers379 treated epoxide 9 with 3 equivalents of mCPBA buffered with Na2HPO4 in an attempt to perform a Barton cascade reaction. It was interesting to note that they isolated only the racemic epoxylactone 10 in 80% yield, but as a single diastereomer. Wipf and coworkers379 also examined additional substrates, including five-and six-membered diosphenol ethers (11380,381 and 13382), and the highly functionalized hydroxy enol ether 15. An epoxide (as found in 9) was clearly not necessary since enone 11 underwent an oxidative rearrangement to afford lactone 12 in 75% yield (Scheme 33). In contrast, the yield for the conversion of the five-membered enone 13 to lactone 14 was only 35%. A carbonyl functionality was also not necessary to facilitate the rearrangement because the epoxy alcohol 15, prepared from the addition of MeLi to ketone 9, succumbed to the same oxidative rearrangement to give hemiacetal 16, albeit in a rather low 20% yield as a racemic single diastereomer.
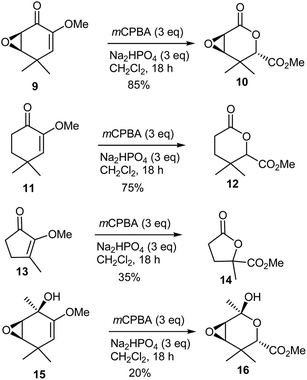 |
| | Scheme 33 Oxidative rearrangement of cyclic enol ethers to α-alkoxyesters with mCPBA. | |
6.4. Oxidative rearrangement of α-alkoxy allenes to α′-alkoxy enones
Lacote and coworkers383 developed an original and rapid access route towards α′-alkoxy enone derivatives by selective oxidation of α-alkoxy allenes with mCPBA (Scheme 34). Both alkyl and silyl ethers of the allene systems underwent efficient migration from C1 to C4 as illustrated in Scheme 34. However, the sterically demanding triisopropylsilyloxy group proved to be too large and this resulted in no rearranged ketone being produced under these conditions. In addition, various combinations of alkyl and aryl groups at C1 viz., R1 and R2 were used in this work (only achiral or racemic allenes were available). This demonstrated that prototropy is the major limitation for migration in α-alkoxy allenes and a secondary factor steering the reactivity away from oxa-cyclization is steric hindrance at C1. Notably, when several oxidizable groups were present on the compound, oxidation occurred exclusively at the allene framework, and not on the allyl, styryl, or propargyl moieties present. Finally, the facile transformational migration of substrates containing protecting groups, such as benzyl, and, above all, trimethylsilyl, provided access to synthetically useful α′-hydroxy enones after deprotection.383
 |
| | Scheme 34 Oxidation of tertiary α-alkoxy allenes to α′-alkoxy enones. | |
In a most intriguing case, a smooth double migration was achieved from the oxidation of the bis-allene 17 (Scheme 35) using 2 equiv. of mCPBA, in which reaction proceeded within 2 h at 0 °C to deliver diketone 18 in 77% yield. No traces of any over oxidation were detected in the product.
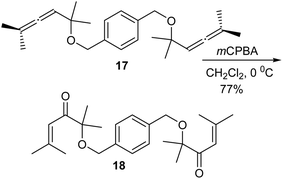 |
| | Scheme 35 Oxidation of a bis-allene derivative. | |
6.5. Miscellaneous rearrangements
An oxidative rearrangement took place during mCPBA epoxidation of the secondary allylic alcohol, auraptenol, leading to the enal shown in Scheme 36. This reaction was used in approaches towards the synthesis of gravol and arnottinin.384–386 The precise reason why the initially formed intermediate epoxide undergoes further rearrangement in this case is not known, although there is the possibility that mCPBA by-product is able to act as an acidic catalyst, initiating protonation of the epoxide oxygen.
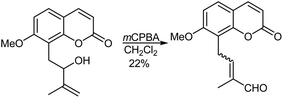 |
| | Scheme 36 mCPBA mediated rearrangement of auraptenol. | |
Ring D in the norsteroidal carboxylic acid chloride 23 did not follow the normal carboxy inversion reaction expected upon treatment with mCPBA (Scheme 37).387,388 Interestingly, it was found that β-acid chloride 19 undergoes an unexpected rearrangement to the allylic cyclopropane 20 in good yield, while the corresponding α-acid chloride 21 gave the expected alcohol 23 in 63% together with the product of an elimination with methyl migration 22 in 37% yield. Conformational analysis of these substrates suggested that the stereochemistry of the acid chloride group dictated the course of rearrangement, since the bond trans to the migrating group must be suitably disposed to participate in a decarboxylation.
 |
| | Scheme 37 Rearrangement of D norsteroidal carboxylic acid chlorides with mCPBA. | |
Williams and coworkers showed that epoxidation of a highly functionalized aryl allene with mCPBA gave a rearranged enone.389 Exposure of 24 to mCPBA resulted in allene oxidation and rearrangement to the enones 25 (dr = 2.3:1, Scheme 38).390 Importantly, there was no evidence of any arene oxidation. Although the precise stereochemical assignment of the major product has not been unequivocally established, the apparent modest selectivity suggests that epoxidation of the disubstituted terminus double bond occurs first, an event which would be expected to take place with high facial selectivity and lead to the predominance of a single allene oxide intermediate (not shown). This stereocenter would be destroyed upon eventual enone formation. The expected high reactivity of the allene oxide toward epoxidation would lead to the rapid formation of a spirodiepoxide. The second epoxidation would be expected to be less selective, thus the stereocenter involved would be retained in the final enone product.
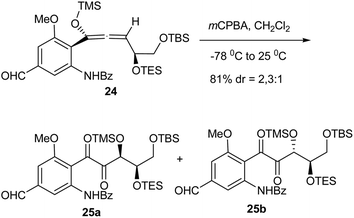 |
| | Scheme 38 Oxidative rearrangement of an aryl allene with mCPBA. | |
7. Cyclization reactions
7.1. One-pot preparation of oxazoles
The oxazole group is one of the key units present in many biologically active natural products, including diazonamides, inthomycins, calyculins and phorboxazoles, and has been extensively used in medicinal chemistry.391–396 Kawano and Togo reported the direct PhI-catalyzed preparation of 2,5-disubstituted and 2,4,5-tri-substituted oxazoles from the reaction between alkyl aryl ketones and nitriles with TfOH and mCPBA (Scheme 39).397 Different iodoarenes such as iodobenzene, 4-iodotoluene, 4-chloroiodobenzene, 4-iodoanisole, 1-iodonaphthalene, 4,4′-diiodobiphenyl, 1,4-bis(4′-iodophenyl)benzene, and poly(4-iodostyrene) were used as catalysts combined with mCPBA and TfOH. However, in the absence of an iodoarene product was not formed at all, for example, in the case of the synthesis of 2-methyl-5-phenyloxazole. However, in the presence of an iodoarene, 2-methyl-5-phenyloxazole could be formed in moderate yield (54–60%) in a one-pot manner, especially when using iodobenzene, 4-iodotoluene, and 4-chloro-iodobenzene. The same reaction could be carried out with butyronitrile and isobutyronitrile instead of acetonitrile using iodobenzene and mCPBA. IL-supported PhI could also be used in the same preparation of oxazoles from ketones and nitriles and could be reused in the same reaction to obtain moderate yields of a variety of oxazoles.397
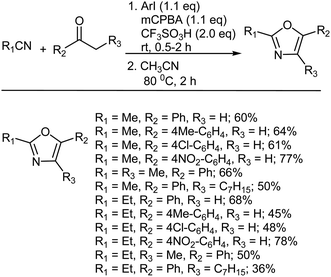 |
| | Scheme 39 Preparation of oxazoles with mCPBA. | |
7.2. One-pot synthesis of tetrahydrobenz[b]azepin-4-ones
Tetrahydrobenz[b]azepin-4-ones represent a biologically significant class of benzene-fused heterocycles and have been studied as mitochondrial benzodiazepine receptor (MBR) antagonists,398 AMPA receptor antagonists,399 and oxytocin and vasopressin antagonists.400 Zhang et al.401 reported a one-pot synthesis of tetrahydrobenz[b]azepin-4-ones from tertiary N-(but-3-ynyl)anilines by using mCPBA (Scheme 40). The aniline nitrogen in the N-(but-3-ynyl)aniline plays an interesting role in relaying “O” from mCPBA to a gold-activated C–C triple bond, which is particularly noteworthy considering that alkynes are generally inert towards mCPBA oxidation under the reaction conditions used (i.e., 0 °C in CH2Cl2).
 |
| | Scheme 40 One-pot synthesis of tetrahydrobenz[b]azepin-4-ones. | |
Interestingly, substituents with different electronic characters were tolerated in the meta and para positions of the aromatic ring, and the use of electron-donating groups such as MeO and Me as well as weakly electron-withdrawing groups such as halides led to better yields than when strongly electron-withdrawing groups were present. In most cases, acceptable to good yields were obtained in this two-step, one-pot transformation. In the case of substrates with meta substituents, the regioselectivity was low. Moreover, in the case of m-NO2, the major product was surprisingly the more hindered ortho-substitution isomer. Noticeably, the presence of functional groups such as halides and NO2 on the benzene ring allows for the easy derivatization of these bicyclic heterocycles when the products are used in further reactions. In addition, a benzyl group was suitable as the aniline nitrogen substituent, and its ready removal opened a good route towards the functionalization of the azepinone nitrogen.401 While longer alkyne chains such as pent-4-ynyl did not present any problems in the transformation, other groups, such as i-Pr or phenyl, when attached at the but-3-ynyl group adversely affected N-oxide formation. Different substituents at the butyne terminus were also studied. The presence of a phenyl group did not lead to any desired product, but the more electron-deficient p-NO2Ph group underwent the reaction in a moderate yield.401
7.3. Cyclization of N-methoxy-2-arylethanesulfonamides
It is well known that sulfonamides possess very useful biological activities.402–409 Cyclic sulfonamides (sultams), in particular, are important as therapeutic compounds410,411 and chiral auxiliaries.412–416 Among these compounds, 3,4-dihydro-2,1-benzothiazine-2,2-dioxides (benzosultams) have proven potent biological activities, such as lipoxygenase inhibitory activity and can be used as drugs for treating heart diseases.417 Hideo and Togo417 reported an ion-supported PhI-catalyzed cyclization of N-methoxy-2-arylethanesulfonamides with mCPBA that formed the corresponding N-methoxy-3,4-dihydro-2,1-benzothiazine-2,2-dioxides in moderate to good yields in the solvent 2,2,2-trifluoroethanol (Scheme 41). In the absence of ion-supported PhI, the product, N-methoxy-3,4-dihydro-2,1-benzothiazine-2,2-dioxide, was not formed at all. The reactive and essential hypervalent iodine intermediate compound, viz., ion-supported [(hydroxy)(tosyloxy)iodo]benzene, is formed in situ and reacts with the N-methoxy-2-arylethanesulfonamide in the presence of mCPBA in an electrophilic manner on the aromatic ring to afford the corresponding N-methoxy-3,4-dihydro-2,1-benzothiazine-2,2-dioxide. Moreover, the ion-supported PhI could be efficiently reused to provide the same products in good yields. The same ion-supported PhI-catalyzed cyclization was carried out on N-methoxy-3-phenylpropionamide and N-methoxy-4-phenylbutyramide using mCPBA to form the corresponding N-methoxy benzo-lactams in moderate yields in 2,2,2-trifluoroethanol.417
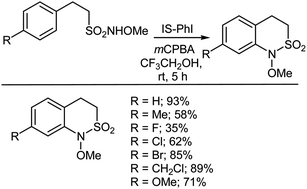 |
| | Scheme 41 Cyclization of N-methoxy-2-phenylethanesulfonamides with mCPBA. | |
7.4. Stereoselective synthesis of 2,5-disubstituted tetrahydrofurans
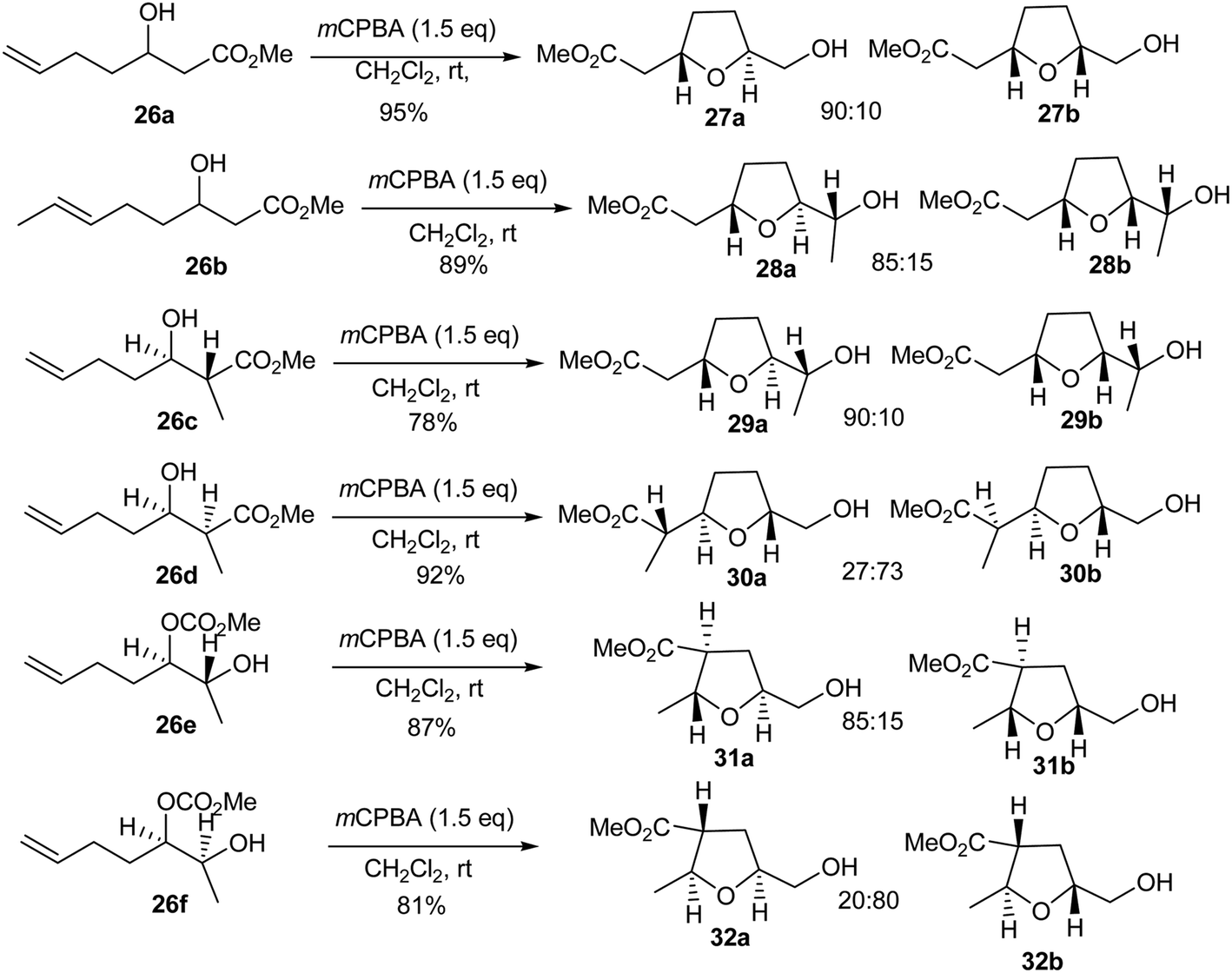
The stereoselective synthesis of 2,5-disubstituted tetrahydrofurans has received considerable attention from organic chemists during the last three decades.418 Iqbal and coworkers418 reported a one pot stereoselective synthesis of cis or trans 2,5-di-substituted tetrahydrofurans in high yields via electrophilic cyclisation of the corresponding cis or trans α- or γ-allyl-β-hydroxyesters mediated by mCPBA (Scheme 42). The allyl β-hydroxyesters 26a–f were treated with mCPBA (1.5 equivalents) in dichloromethane for 4–6 hours and each afforded a single diastereomer of a 2,5-disubstituted tetrahydrofuran as the major product. The β-hydroxy ester 26a predominantly provided 27a, whereas 26b underwent a smooth cyclisation to yield 28a as the major product. Similarly, trans β-hydroxyester 26c provided the trans–cis isomer 29a as the major product in excellent yield. Conversely, the corresponding cis diastereomer 26d provided the cis–trans isomer 30b as the major product, the trans diastereomer of α-allyl-β-hydroxy ester 26e was smoothly cyclised, giving the trans–trans tetrahydrofuran 31a as the major product. In contrast, the corresponding cis diastereomer 26f cyclised very slowly and provided the cis–trans diastereomer 32b as the major product.418
 |
| | Scheme 42 Synthesis of tetrahydrofurans with mCPBA. | |
The high cis-stereoselectivity during formation of tetrahydrofurans 27a–29a can be explained by invoking the involvement of the methoxycarbonyl group during the 5-exo-trig cyclisation process. This assumption is based on the fact that the electrophilic cyclisation of γ,δ-unsaturated alcohols usually results in trans stereochemistry at the ring junction. In this case, the alcohols 26a–c mainly give rise to the cis stereochemistry at the ring junction, indicating that the methoxycarbonyl group may be responsible for this specific stereoselectivity. On the other hand, the cis stereochemistry observed during the electrophilic cyclisation of alcohols 26a–c could be explained by a stabilising interaction between the developing positive charge on the protonated oxirane and the oxygen lone pair of the ester carbonyl via hydrogen bonding, which would most likely be responsible for the high stereoselectivity.418
7.5. Spirocyclization
A great variety of spirocyclic compounds are known to exist in nature and several classes of these compounds have received significant interest in recent years as privileged structures found in pharmaceuticals, organic materials for optoelectronics and other applications, chiral ligands and catalysts for use in synthesis, to mention but a few examples.419–424 Kita and coworkers425 developed a very effective one pot spirocyclization procedure for installing nucleophiles (Nu = N3, NO2, SCN, SO2Tol, and halogens) via iodonium(III) salts using a combination of an iodoarene and mCPBA (Scheme 43). This procedure involves three key steps: firstly, in situ generation of a cationic hypervalent iodine species from the iodoarene using mCPBA; secondly, alkyne activation by electrophilic iodine, which induces ipso-cyclization of the aryl alkynes as directed by the methoxy group on the aryl ring, forming a spirocyclized iodonium(III) salt; and thirdly, the installation of a nucleophile via a reductive coupling with the previously formed salt to produce the functionalized spirocyclic compound.425
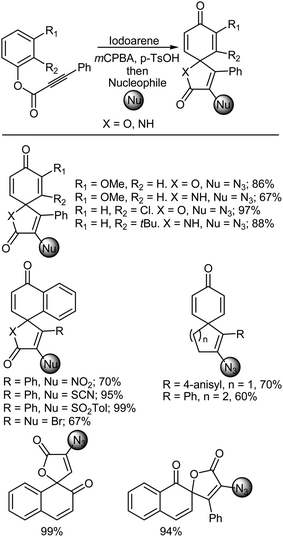 |
| | Scheme 43 Spirocyclization with mCPBA. | |
Regarding the iodoarene, an ortho-diiodinated biaryl structure was essential for developing transformations that showed good performance. Minor manipulations of the substituents in the bis(iodoarene) contributed to, not only the ipso-cyclization and formation of the spirocyclized iodonium salt, but also the final substitution event with the nucleophile at the alkenyl moiety. In addition to the construction of five- and six-membered para-spirocyclized cyclohexadienones, the application of this protocol to the formation of valuable ortho-spirolactone structures that are frequently seen in natural products426–429 was also successful based on the demonstrations conducted on the aryl alkynes. In addition to azide functionality, a series of nitrogen, sulfur, and halogen nucleophiles effectively participated in the spirocyclization using starting materials that contained different alkynes.
7.6. Oxidative cyclization of aromatic nuclei
Phenanthrene ring and biaryl linkages are found extensively in natural products, pharmaceuticals and many other important organic molecules,430–432 especially in the phenanthroindolizidine and phenanthroquinolizidine alkaloids and 1,1′-binaphthalene derivatives. Phenanthroindolizidine and phenanthroquinolizidine alkaloids exhibit interesting pharmacological properties, among which antitumor activity is the most notable.432–434 Wang and coworker435 reported that readily available and non-toxic FeCl3 was able to catalyze an intramolecular oxidative coupling for the direct construction of an phenanthrene ring using mCPBA as the sole oxidant at room temperature and in excellent yield (Scheme 44). Initially, the intramolecular oxidative coupling (cyclization) reaction of (E)-methyl-2,3-bis(3,4-dimethoxyphenyl)acrylate (33a) was investigated. The desired coupling product 33b was obtained in 99% yield using FeCl3 as the catalyst and mCPBA as the oxidant. When compared with di-tert-butylperoxide (DTBP), mCPBA demonstrates a higher oxidative ability. Further investigations showed that mCPBA alone was not effective for the oxidative coupling of 33a. Similarly, substrate (E)-34a, which also had an electron-withdrawing group (–CO2H) on the double bond, was found to react smoothly under the same conditions, giving the desired product 34b in 96% yield. The intramolecular oxidative coupling of (Z)-35a and (Z)-36a with FeCl3 (10 mol %) and mCPBA also gave the corresponding coupling products 33b and 36b, respectively, in almost quantitative yields, suggesting that the configuration of the double bond has no effect on the outcome of the oxidative reaction.
 |
| | Scheme 44 Intramolecular oxidative cyclization of diphenyacrylates. | |
8. Miscellaneous
8.1. α-Functionalization
8.1.1. Non-asymmetric (racemic) α-oxytosylation of ketones. α-Tosyloxy ketones are very important strategic precursors for the construction of various heteroaromatics, such as thiazoles, imidazoles, imidazo[1,2-a]pyridines, oxazoles, selenazoles, pyrazoles, and benzofurans.436–453 Interestingly, method A [PhI (0.1 eq.), mCPBA (1.1 eq.), RSO3H·H2O (1.1 eq.)],454,455 method B [polymer-supported-PhI (0.1 eq.), mCPBA (2.1 eq.), TEMPO (cat.), p-TsOH·H2O (1.1 eq.)],456 method C [I2 (0.1 eq.), mCPBA (2.2 eq.), p-TsOH·H2O (2.1 eq.)],457 method D [I2 (0.1 eq.), mCPBA (2.2 eq.), t-BuPh (0.2 eq.), p-TsOH·H2O (1.5 eq.)]457 and method E [IL-supported-PhI (0.1 eq.), mCPBA (1.3 eq.), IL (0.1 eq.), p-TsOH·H2O (1.1 eq.)]458 could all be used to selectively convert ketones to their corresponding α-tosyloxyketones (Scheme 45).
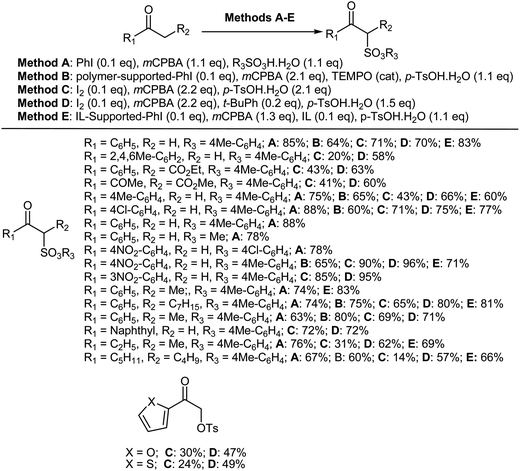 |
| | Scheme 45 α-Tosyloxylation of ketones. | |
Various ketones viz., alkyl aryl ketones, dialkyl ketones and cyclic ketones, were converted to the corresponding α-tosyloxyketones using methods A–E. Aldehydes were also reacted using the conditions of method A. However, the corresponding α-tosyloxyaldehydes were not obtained due to the instability of the products under the reaction conditions. In unsymmetrical methyl ketones, α-tosyloxylation at the alkyl group was favored over the methyl group when using method A. However, the yields of α-tosyloxy ketones produced from alkyl aryl ketones bearing an electron-donating group on the aromatic ring were low when using method C, and those of dialkyl ketones were also low. Overall, the yields reported using method D were higher than those using method C. Notably, α-tosyloxyketones were not obtained at all in the absence of iodine using methods C and D. Moreover, the ionic-liquid reaction media containing a catalytic amount of IL-supported PhI that was used in method E could be reused for the α-tosyloxylation of ketones.
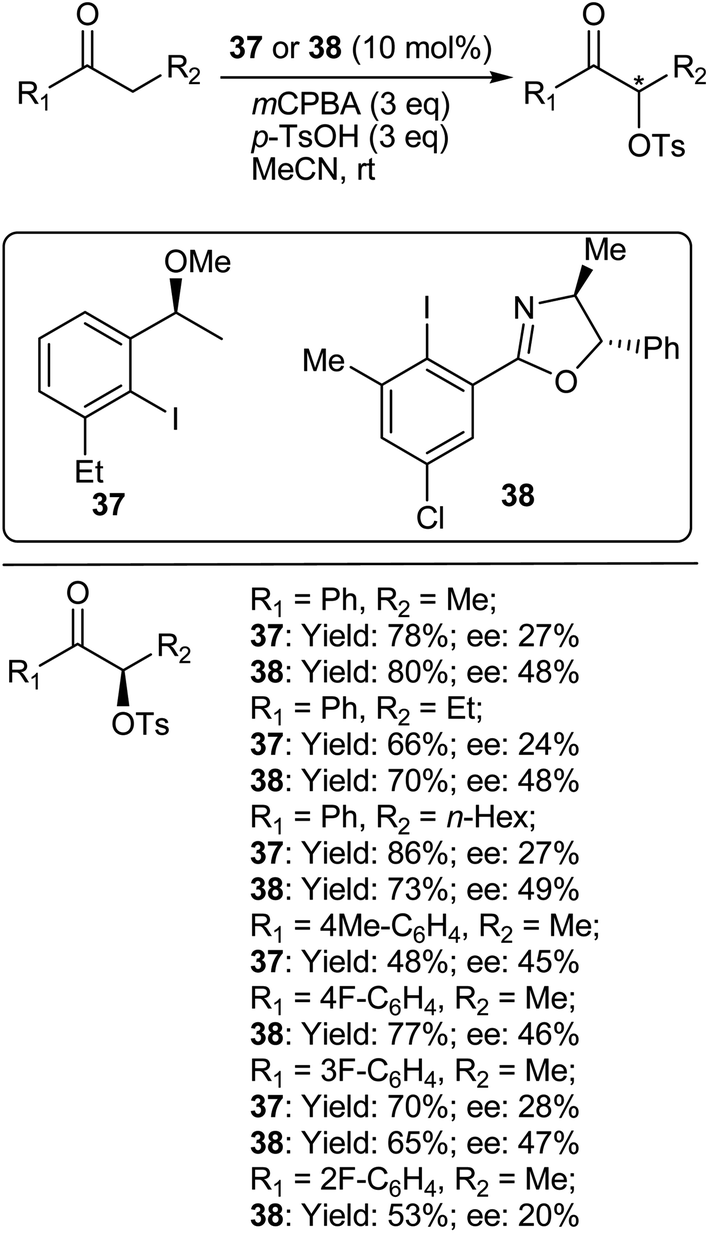
8.1.2. Asymmetric α-oxytosylation of ketones. Wirth and coworkers459,460 reported the next obvious development viz., the enantioselective α-oxytosylation of ketones, using mCPBA as a stoichiometric oxidant and p-toluenesulfonic acid monohydrate (TsOH·H2O) as the source of the tosylate nucleophile (Scheme 46). The reactions were performed using non-stoichiometric (10 mol%) quantities of iodoarene 37 and simple propiophenone derivatives generally underwent reaction in good yields and with reliable enantioselectivities. Electron-rich propiophenones underwent side reactions, possibly as a result of their anisole function groups,461 and could not be purified. Increasing the steric congestion on the prochiral methylene group led to a slower reaction. Using indanone resulted in a high yield and enantioselectivity consistent with the acyclic propiophenone derivatives, but tetralone underwent a very slow reaction giving an almost racemic product. However, cyclopentanone and cyclohexanone underwent the Baeyer–Villiger oxidation under these conditions.
 |
| | Scheme 46 Asymmetric α-oxytosylation of ketones. | |
In another study, Legault and coworkers462 explored the reaction scope with catalyst 38 and treated a variety of ketones under the same previously described conditions. Variation of the alkyl chain of the propiophenone did not result in any significant differences in terms of the yield or enantioselectivity. Use of a cyclic ketone, such as indanone, resulted in a decrease in selectivity. Surprisingly, tetralone was unreactive under the reaction conditions. The method tolerated electron-withdrawing groups on the aryl ring of propiophenone fairly well, but the presence of an electron-donating groups resulted in a drastic decrease in reactivity, with the p-methoxy derivative being almost unreactive. Introduction of ortho substitution on the aryl ring of the propiophenone also had a detrimental effect on both the yield and selectivity. Finally, aliphatic ketones were tested and found to be unreactive under the reaction conditions.
8.1.3. α-Acetoxylation of ketones. Ochiai and coworkers463 showed that exposing acetophenone to dried mCPBA (1.4 equiv.) in acetic acid in the presence of a catalytic amount (10 mol%) of iodobenzene, BF3·Et2O (3 equiv.), and water (5 equiv.) at room temperature provided α-acetoxyacetophenone in 84% yield (Scheme 47). It is important to note that the addition of water was crucial to the success of the α-acetoxylation of acetophenone since in the absence of water, α-oxidation was almost inhibited with 95% recovery of the ketone. Formation of α-acetoxyacetophenone was also not observed when the reaction was carried out in the absence of iodobenzene. Similarly, BF3·Et2O was also an essential contributor to the direct α-oxidation of acetophenone and α-acetoxylation did not take place at all in the absence of BF3·Et2O. The use of other Lewis acids instead of BF3·Et2O, such as Yb(OTf)3, CF3SO3H, and HBF4·Me2O, afforded only a moderate yield of α-acetoxyacetophenone.463
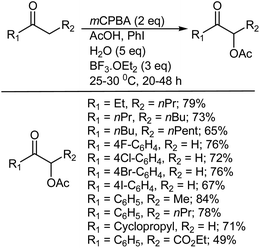 |
| | Scheme 47 α-Acetoxylation of ketones. | |
A variety of dialkyl and alkyl aryl ketones were also smoothly oxidized at the α-position under these catalytic conditions and afforded α-acetoxy ketones in good yields. Substituted acetophenones with halogens (F, Cl, Br, and I) at the para position gave comparable results, indicating that mCPBA was highly selective for the oxidation of iodobenzene over p-iodoacetophenone. In the case of unsymmetrical ketones, the oxidation of a methylene group in linear long chain alkyl groups was favored over that of a methyl group, indicating that the reactivity of α-methylene groups toward acetoxylation decreases in the order ethyl > pentyl > nonyl.463
8.2. Diels–Alder reactions
Ethyl propiolate undergoes a one-pot three-step thioconjugate addition–oxidation–Diels–Alder cycloaddition when treated with a variety of thiols in the presence of a catalytic base, mCPBA, lithium perchlorate and cyclopentadiene (Scheme 48).464 Electron-rich aryl thiols were found to be the most successful substrates. In some cases, addition of a second equivalent of LiClO4 during the cycloaddition step was unnecessary to achieve both high yield and selectivity. In the case of p-bromothiophenol, the reaction was performed in 1,2-dichloroethane in order to achieve a higher reflux temperature during the oxidation step, thus ensuring full oxidation to the sulfone. In general, halogenated thiophenol derivatives appear to react somewhat less selectively than their counterparts, which results in lower isolated yields of the major cycloaddition adduct. Benzyl mercaptan reacted analogously to the S-aryl thiols, providing the major isomer in 67% yield. Diastereoselectivity varied somewhat from substrate to substrate and both the exo isomer derived from the Z enoate and diastereomers resulting from the cycloaddition of the E enoate were frequently observed as minor products, but in all cases the major endo isomer was easily purified using column chromatography.464
 |
| | Scheme 48 One-pot three-step Diels–Alder reactions of aryl thiols with mCPBA. | |
In another study, Thiemann et al.465 reported that brominated anthraquinones could be synthesized directly from bromothiophenes via a Diels–Alder reaction with 1,4-naphthoquinones in the presence of mCPBA (Scheme 49). Under these conditions, cycloaddition takes place between a thiophene S-oxide intermediate and the 1,4-naphthoquinone, after which the primary sulfoxy-bridged cycloadduct formed loses its SO bridge with concomitant aromatization.
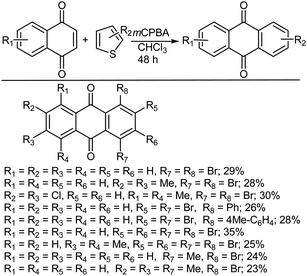 |
| | Scheme 49 Synthesis of brominated anthraquinones with mCPBA. | |
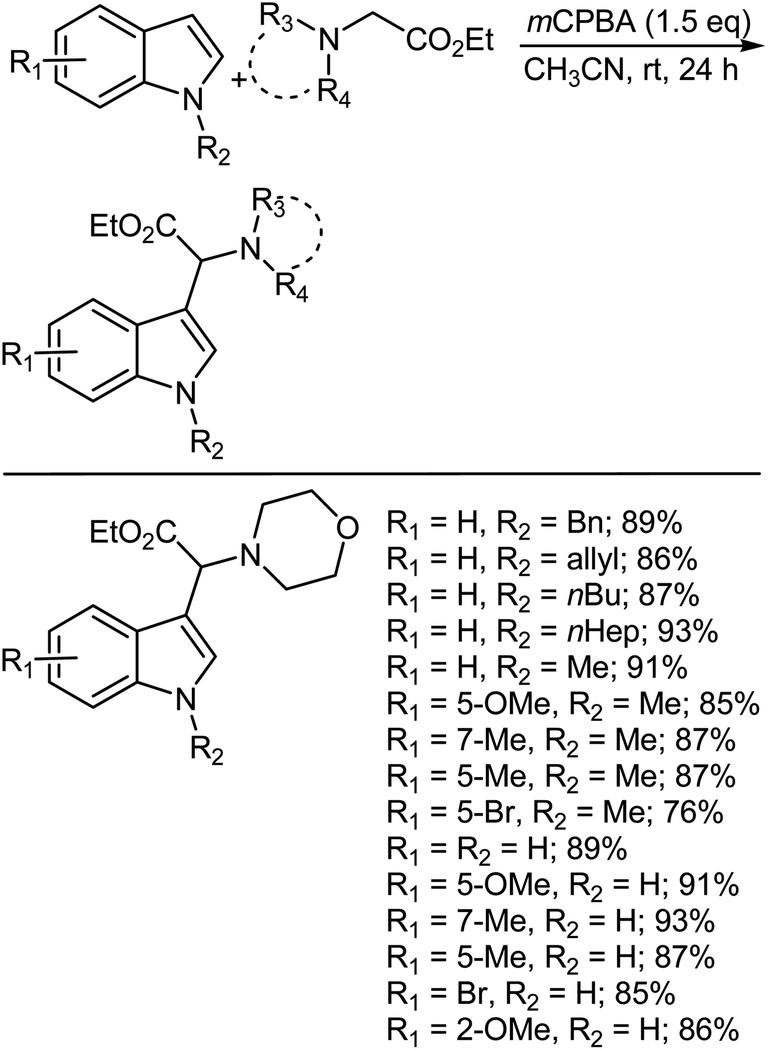
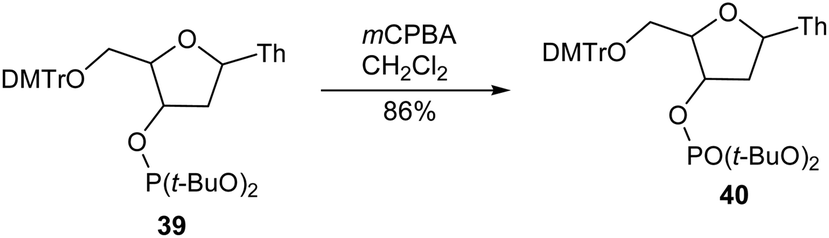
8.3. Oxidative coupling of indoles with ethyl 2-(disubstituted amino)acetates
Indolylglycine derivatives are important synthetic intermediates or building blocks for drug development466 as well as natural product synthesis.467–472 Bao and coworkers found that the oxidative coupling of ethyl 2-(disubstituted amino)acetates with indoles proceeded in the presence of mCPBA under metal-free conditions to provide indolylglycine derivatives in satisfactory to excellent yields (Scheme 50).473 mCPBA acted as an oxidant in this methodology. Different solvents were initially tested, but CH3CN proved to be the best. The reactions of different indole derivatives with various N-protecting groups such as benzyl, allyl, n-butyl, n-heptyl, and methyl proceeded smoothly with ethyl 2-morpholinoacetate and furnished the corresponding coupling products in good to excellent yields (86–93%). These results indicated that the size of the N-protecting group did not influence the reactivity of the indole substrate. N-Me indoles bearing electron-donating groups such as OMe and Me on their benzene rings are also able to undergo the desired oxidative coupling reaction smoothly to yield the corresponding products in good yields (85–87%). However, the N-Me indole bearing a bromine atom, an electron-withdrawing group, on the benzene ring showed relatively low reactivity in this type of oxidative coupling reaction. These results showed that the reaction yield was indeed influenced by the electronic properties of the substituent linked to the benzene ring of the indole moiety. Subsequent studies revealed that the free (NH)-indole substrates could also undergo this type of oxidative coupling reaction.473
 |
| | Scheme 50 Oxidative coupling of various indole derivatives with ethyl 2-morpholinoacetate. | |
8.4. Oxidative cleavage of polycyclic systems
Medium- to large-sized rings can be synthesized via the oxidative cleavage of internal double bonds in polycyclic systems. Interestingly, nine- and ten-membered ring containing compounds that were the result of mCPBA-mediated oxidative cleavage reactions were shown to exhibit atropisomerism.474 The scope of this oxidative cleavage reaction was explored as a function of the substituents on ring A of the substrate shown in Scheme 51. In general, the yield decreased slightly in the presence of substituents at the C9-, C10- and C11-positions in the series of reactants where n = 1, but the reactions proceeded in moderate to good yield. When an electron-donating R2 substituent was incorporated onto ring E, the reaction failed entirely, with only extensive decomposition being observed (Scheme 51). The presence of either an electron-withdrawing or electron-donating substituent in ring A was unable to rescue the reaction outcome when an electron-donating substituent was included on ring E. However, the expansion of ring C in the reactant was well tolerated.474
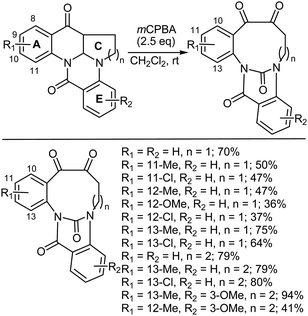 |
| | Scheme 51 Oxidative cleavage reaction with mCPBA. | |
8.5. Heteroatom oxidation
8.5.1. Oxidation of sulfides to sulfoxides and/or sulfones. Sulfones are valuable synthetic intermediates for the construction of chemically and biologically important molecules475–478 especially those that have demonstrated biological activities.479–486 n-Butanethiol was oxidized with mCPBA in CH2Cl2 at −30 °C to afford n-butanesulfinic acid (n-BuSO2H) in 82% yield and, interestingly, other thiols reacted in a similar fashion.487 Sulfides could be oxidized chemoselectively to sulfoxides using mCPBA at −70 °C.488 Three reagents viz., mCPBA, sodium periodate and iodosylbenzene are regarded as ideal for the oxidation of sulfides to sulfoxides.489 mCPBA is the most popular and extensively used reagent for the oxidizing sulfides to their corresponding sulfoxides and sulfones and a selection of results is summarized in Scheme 52.490–549
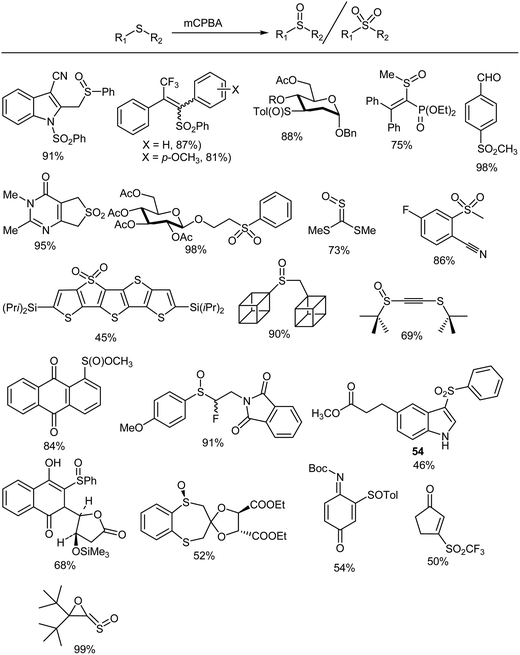 |
| | Scheme 52 Oxidation of sulfides to sulfoxides or sulfones with mCPBA. | |
8.5.2. Oxidation of phosphorous. mCPBA was used to stereospecifically oxidized phosphate 39, affording phosphate 40 in 86% yield (Scheme 53).550 Similarly, the mCPBA oxidation of thiophosphate triesters provides the corresponding phosphate esters with retention of configuration.551 In another study, Chow and Berkman552 prepared phosphate esters of N-phosphoryl amino acids by using mCPBA to oxidize the phosphorous.
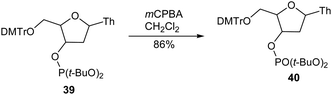 |
| | Scheme 53 An example of phosphorous oxidation with mCPBA. | |
8.5.3. Oxidation of selenides. The selenoyl group has been demonstrated to be a good leaving group, therefore, its generation within a compound can lead to the preparation of cyclic compounds if other functional groups are correctly predisposed. In this way, oxazoline 42 was prepared through the oxidation of selenide 41, followed by treatment of the oxidized material with base.553 Similarly, the oxidation of selenide 43 with mCPBA was very efficient, introducing exocyclic unsaturation under mild conditions for the preparation of compound 44.554 Notably, phenyl selenides react rapidly with mCPBA at −10 °C, but at higher temperatures of between 0 °C to rt they are inclined to undergo a cis elimination (Scheme 54).555
 |
| | Scheme 54 Oxidation of selenides with mCPBA. | |
8.6. Oxidation of C–H bonds
The selective functionalization of saturated hydrocarbons is of major importance in synthetic organic chemistry and over the past few decades considerable attention devoted to the development of efficient methods for both regio- and stereoselective C–H bond activation.556 Kim et al.557 reported the catalytic hydroxylation of aliphatic hydrocarbons by mCPBA in the presence of electron-deficient iron(III) porphyrin complexes (Scheme 55). High yields of alcohol products were obtained together with minor amounts of ketones. The hydroxylation of cis- and trans-1,2-dimethylcyclohexane provided the corresponding tertiary alcohols with high stereoretention, indicating that these reactions are highly stereospecific. Hydroxylation of norbornane provided the exo-norborneol as a major product. In another study, Konoike et al.558 reported a novel allylic hydroxylation of triterpenes using a mCPBA–Fe(PFPP)Cl mixture. It was apparent that the reactions were catalyzed by the modality of Fe(PFPP)Cl because oleanolic acid, ursolic acid and dihydrolanosterol as well as their derivatives bearing sterically hindered olefin groups were all converted to the corresponding allylic alcohols under relatively mild conditions.
 |
| | Scheme 55 Oxidation of C–H bonds with mCPBA. | |
8.7. Generation of aryl iodide(III) compounds from iodoarenes
Hypervalent iodine compounds have a long history of being widely used in organic synthesis.559 Kita et al. demonstrated the synthesis of novel 1,3,5,7-tetrakis[4-(diacetoxy-iodo)phenyl]adamantane and tetrakis[4-(diacetoxy-iodo)phenyl]methane from 1,3,5,7-tetrakis(4-iodophenyl)adamantane and tetrakis(4-iodophenyl)methane, respectively, using mCPBA under diluted conditions at room temperature.83 Generally, room temperature was required for the synthesis of the [hydroxy(sulfonyloxy)iodo]arenes from the reaction of the corresponding (diacetoxyiodo)arenes with p-toluenesulfonic acid monohydrate, due to the high reactivity of the [hydroxy(sulfonyloxy)iodo]arenes. Yamamoto and Togo560 showed that a variety of [hydroxy(sulfonyloxy)iodo]arenes could be efficiently obtained in high yields from the reaction of the relevant iodoarene with mCPBA in the presence of a sulfonic acid in a small amount of chloroform at room temperature, through a one-pot procedure. Various sulfonic acids, including p-toluenesulfonic acid monohydrate, were reacted to furnish the corresponding [hydroxy(sulfonyloxy)iodo]benzenes in good yields (Scheme 56). 1-(Arenesulfonyloxy)benziodoxolones could also be prepared from o-iodobenzoic acid in a one-pot procedure.
 |
| | Scheme 56 One-pot preparation of [hydroxy(sulfonyloxy)iodo]arenes from iodoarenes with mCPBA. | |
In another study, Olofsson et al.561 reported the one-pot synthesis of neutral and electron-rich [hydroxy(tosyloxy)iodo]arenes from iodine and arenes, thereby avoiding the need for expensive iodine(III) precursors (Scheme 57). tert-Butylbenzene, p-xylene and mesitylene proved to be excellent substrates, delivering the corresponding products in good yields. On the other hand, biphenyl was surprisingly unreactive, and even a prolonged reaction time failed to improve the yield. The acid used was subsequently varied with benzene as the arene. Methanesulfonic acid, 2-naphthalenesulfonic acid, and benzenesulfonic acid all delivered the corresponding [hydroxy(tosyloxy)iodo]arenes in good yields, whereas camphorsulfonic acid did not work.
 |
| | Scheme 57 Synthesis of electron-rich [hydroxy(tosyloxy)iodo]arenes. | |
8.8. Oxylactonization of ketocarboxylic acids
Ishihara et al.562 reported on the hypervalent iodine-catalyzed oxylactonization of ketocarboxylic acids to ketolactones in the presence of iodobenzene (10 mol%), p-toluenesulfonic acid monohydrate (20 mol%) and mCPBA as a stoichiometric co-oxidant (Scheme 58). 1-Napthyl and 2-napthyl ketones afforded the corresponding ketolactones in good yields. In contrast, 2-methoxyphenyl ketone was transformed to the corresponding lactone in only 41% yield, with Baeyer–Villiger oxidation products obtained as the alternative main products. Unfortunately, picolinoyl lactone and a six-membered δ-lactone were obtained in only 38% and 15% yield, respectively.
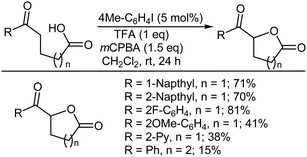 |
| | Scheme 58 In situ-generated hypervalent iodine-catalyzed oxylactonization of ketocarboxylic acids. | |
8.9. Oxidative ring opening of 1,3-diarylbenzo[c]heterocycles
Mohanakrishnan et al.563 reported on the oxidative cleavage of benzo[c]heterocycles using mCPBA and the reaction of 1,3-diaryl benzo[c]heterocycles with mCPBA at room temperature over 5 min, which led to the formation of 1,2-diaroylbenzenes in good to excellent yields (Scheme 59). In the case of 1,3-dithienylbenzo[c]thiophene, the mCPBA-mediated oxidative cleavage provided an unsymmetrical diketone formed through the oxidation of a thiophene unit into an S,S-dioxide moiety. However, the reaction of diarylbenzo[c]thiophenes provided the respective diketones in excellent yields. The oxidative ring-opening reaction of 1,3-diarylbenzo[c]furans proceeded with relatively better yields than their respective benzo[c]thiophenyl heterocycles. Diarylbenzo[c]selenophenes also underwent oxidative cleavage to furnish the corresponding diketones in slightly reduced yields. In the case of thiophene-tethered benzo[c]furans, oxidation of the thiophene unit was not observed. As expected, the benzo[c]furans linked to dihexylfluorene, pyrene, and diphenylmethane groups also underwent oxidative cleavage affording their respective diketones in excellent yields.563
 |
| | Scheme 59 Oxidative ring opening of 1,3-diarylbenzo[c]heterocycles. | |
8.10. Oxidation of indoles
Peracids have been widely used for the oxidation of 2,3-disubstituted indoles.564 Hino demonstrated that treatment of tetrahydrocarbazole with mCPBA at −60 °C in CH2l2 provided hydroxy-4aH-carbazole as the major product.565 Similarly, the hydroxyindolenine could be oxidized in good yield to the corresponding ketoamide using mCPBA in the presence of H2SO4.565 On the other hand, mCPBA oxidation of aristoteline (a piperidino-indole alkaloid) at −40 °C provided the corresponding hydroxyindolenine in 94% yield. However, at 25 °C, the hydroxyindolenine was obtained as the major product (57%), but with a 21% yield of the corresponding ketoamide.566 Furthermore, ketoamides have been synthesized by mCPBA oxidation of tetrahydrobenzo[b][1,8]naphthyridin-5(7H)-one567 and N-methylazetopyridoindoles.568 Inspired by these results, Husson et al.569 reported on the oxidative cleavage of an indole 2,3 double bond according to the experimental conditions reported by Kurihara568 (mCPBA, room temperature in CH2Cl2) for N-substituted indole δ-lactones 45a–c. However, the expected ketoamides were not obtained and new heterocycles were formed instead, to which the structures of 4,5-dihydrospiro[furan-3(2H),3′-indole]-2,2′-(1′H)-diones 46a–c were attributed (Scheme 60).
 |
| | Scheme 60 Oxidation of indoles with mCPBA. | |
8.11. Miscellaneous transformations
Góra et al.570 reported on the oxidation of naphthylcycloalkenes with mCPBA followed by the acid catalyzed rearrangement of either the diol or epoxide, and provided a series of naphthylcycloalkanones in a very simple manner (Scheme 61). The conditions allowed for preparation of naphthylcycloalkanones on a multigram scale. Initially, most of the substituted naphthylcycloalkenes gave only 10–20% yields of the target naphthylcycloalkanones when using H2O2, but changing the conditions to mCPBA gave more satisfactory yields.
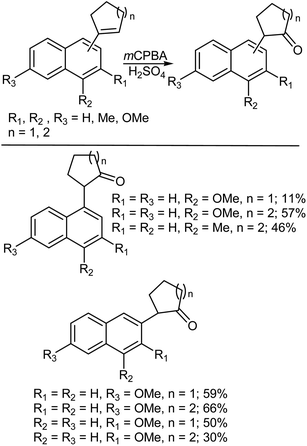 |
| | Scheme 61 Oxidation of naphthylcycloalkenes with mCPBA. | |
Kim et al.571 demonstrated an efficient conversion of cyclic acetals to their corresponding hydroxy alkyl esters in good to excellent yields through oxidation using mCPBA (Scheme 62). Interestingly, all of the cyclic acetals evaluated provided hydroxy alkyl esters without any problems. From these results, it was suggested that aldehydes could also be converted to the corresponding hydroxy alkyl esters. Besides symmetrical cyclic acetals, unsymmetrical cyclic acetals were also converted to hydroxy alkyl esters. It is noteworthy that the unsymmetrical cyclic acetals afforded only one isomer as the product.
 |
| | Scheme 62 Conversion of cyclic acetals into their corresponding hydroxy alkyl esters. | |
Pinnick et al.572 reported the synthesis of amides derived from thioamides using mCPBA. This reaction was quite rapid and occurred at the total exclusion of olefin epoxidation of any side chain olefins when present (Scheme 63). Furthermore, this methodology could be applied to both thioamides and thiolactams, proceeding in high yield. Primary, secondary and tertiary thioamides undergo this transformation with equal efficiency. For example, butyramide, aprolactam, and N-methylpyrrolidone were produced in 76%, 89%, and 82% yield, respectively, from the relevant thioamide precursors.
 |
| | Scheme 63 Synthesis of amides from thioamides. | |
Inokuchi and Kawafuchi573 reported that O-benzyl- and O-allyl-TEMPOs could be transformed to the corresponding carbonyl compounds by using mCPBA (Scheme 64). Different oxidizing reagents viz., Mn(OAc)3, Cu(OAc)2 and tert-BuOOH were examined, but it turned out that mCPBA was the most efficient oxidant, causing the most rapid reaction at 0–5 °C and affording the expected products in excellent yields.
 |
| | Scheme 64 Conversion of O-benzyl- and O-allyl-TEMPOs to carbonyl compounds. | |
9. Summary
The different synthetic methods discussed in this review show that mCPBA is a versatile reagent for use in organic synthesis. mCPBA is a cheap, commercially available oxidant that easily oxidizes numerous functional groups. It is an efficient single oxygen atom donor because it contains a non-symmetrical O–O bond, which is heterolytically cleaved during the oxidation cycle. The tactical utilization of mCPBA in synthetic plans can help to replace tedious organic transformations with simpler routes. One drawback of mCPBA is that this oxidizing reagent will only epoxidize electron-rich olefins and allylic or homoallylic alcohols and it also requires a directing group. One other drawback, which needs to be mentioned, is that a relatively large excess of mCPBA may be required in some reactions in order to consume all of the starting material. It is interesting to note that, a major factor militating against this is that mCPBA can be reused when it is in stoichiometric excess. Owing to the discovery of a variety of novel applications, mCPBA is becoming an increasingly important reagent in synthetic organic chemistry. We hope that this review may act as a catalyst for boosting the application of mCPBA in organic synthesis.
References
- M. Hudlicky, Oxidations in Organic Chemistry, ACS Monograph Series 186, American Chemical Society, Washington, DC, 1990 Search PubMed.
- Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1st edn, 1991, vol. 7 Search PubMed.
- R. A. Sheldon, in Catalytic Oxidation, ed. R. A. Sheldon and R. A. van Santen, World Scientific, Singapore, 1995, p. 239 Search PubMed.
- J. H. Clark and D. J. Macquarrie, Org. Process Res. Dev., 1997, 1, 149 CrossRef CAS.
- W. F. Hoelderich and F. Kollmer, Pure Appl. Chem., 2000, 72, 1273 CrossRef CAS.
- T. Dohi and Y. Kita, Chem. Commun., 2009, 2073 RSC.
- S. Cha, J. Hwang, M. G. Choi and S. K. Chang, Tetrahedron Lett., 2010, 51, 6663 CrossRef CAS PubMed.
- S. Kamijo, S. Matsumura and M. Inoue, Org. Lett., 2010, 12, 4195 CrossRef CAS PubMed.
- E. Gipstein, F. Nichik and J. A. Offenbach, Anal. Chim. Acta, 1968, 43, 129 CrossRef CAS.
- R. Tank, Synlett, 2007, 664 CrossRef CAS PubMed.
- R. N. McDonald, R. N. Steppel and J. E. Dorsey, Organic Syntheses, Coll., 1988, 6, 276 CAS; R. N. McDonald, R. N. Steppel and J. E. Dorsey, Organic Syntheses, Coll., 1970, 50, 15 Search PubMed.
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943 CrossRef CAS PubMed.
- X. Zhang, A. Hu, C. Pan, Q. Zhao, X. Wang and J. Lu, Org. Process Res. Dev., 2013, 17, 1591 CrossRef CAS.
- G. R. Krow, Org. React., 1993, 43, 251 CAS.
- M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737 CrossRef CAS.
- M. Harmata and P. Rashatasakhon, Tetrahedron Lett., 2002, 43, 3641 CrossRef CAS.
- G. Mehta and N. Mohal, J. Chem. Soc., Perkin Trans. 1, 1998, 505 RSC.
- H. R. Bjørsvik, G. Occhipinti, C. Gambarotti, L. Cerasino and V. R. Jensen, J. Org. Chem., 2005, 70, 7290 CrossRef PubMed.
- F. Grein, A. C. Chen, D. Edwards and C. M. Crudden, J. Org. Chem., 2006, 71, 861 CrossRef CAS PubMed.
- R. Sandaroos, M. T. Goldani, S. Damavandi and A. Mohammadi, J. Chem. Sci., 2012, 124, 871 CrossRef CAS.
- C. M. Crudden, A. C. Chen and L. A. Calhourn, Angew. Chem., Int. Ed., 2000, 39, 2851 CrossRef CAS.
- S. Yamabe and S. Yamazaki, J. Org. Chem., 2007, 72, 3031 CrossRef CAS PubMed.
- P. A. Smith, in Molecular Rearrangements, ed. P. de Mayo, Interscience, New York, 1963; Vol. 1 Search PubMed.
- R. Criegee, Justus Liebigs Ann. Chem., 1948, 560, 127 CrossRef CAS.
- L. Zhou, X. Liu, J. Ji, Y. Zhang, X. Hu, L. Lin and X. Feng, J. Am. Chem. Soc., 2012, 134, 17023 CrossRef CAS PubMed.
- C. Bolm, in Asymmetric Synthesis: The Essentials, ed. M. Christmann, S. Braese, Wiley-VCH, Weinheim, Germany, 2006, p. 57 Search PubMed.
- C. Bolm and O. Beckmann, in Comprehensive Asymmetric Catalysis, ed. E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Springer, Berlin, 1999, vol. 2, p. 803 Search PubMed.
- C. Bolm, in Advances in Catalytic Processes, M. P. Doyle, JAI Press, Greenwich, CT, 1997, vol. 2, p. 43 Search PubMed.
- C. Bolm, G. Schlingloff and K. Weickhardt, Angew. Chem., Int. Ed. Engl., 1994, 33, 1848 CrossRef.
- A. Gusso, C. Baccin, F. Pinna and G. Strukul, Organometallics, 1994, 13, 3442 CrossRef CAS.
- C. S. Penkett and I. D. Simpson, Tetrahedron Lett., 2001, 42, 3029 CrossRef CAS.
- H. T. Nagasawa, J. G. Kohlhoff, P. S. Fraser and A. A. Mikhail, J. Med. Chem., 1972, 15, 483 CrossRef CAS.
- I. A. O'Neil, E. Cleator and D. J. Tapolczay, Tetrahedron Lett., 2001, 42, 8247 CrossRef CAS.
- R. E. Sammelson and M. J. Kurth, Tetrahedron Lett., 2001, 42, 3419 CrossRef CAS.
- G. M. Rubottom, M. A. Vazquez and D. R. Pelegrina, Tetrahedron Lett., 1974, 15, 4319 CrossRef.
- G. M. Rubottom and J. M. Grube, J. Org. Chem., 1978, 43, 1599 CrossRef CAS.
- A. Hassner, R. H. Reuss and H. W. Pinnick, J. Org. Chem., 1975, 40, 3427 CrossRef CAS.
- A. G. Brook and D. M. MAcrae, J. Organomet. Chem., 1974, 77, C19 CrossRef CAS.
- C. R. Krueger and E. G. Rochow, J. Organomet. Chem., 1964, 1, 476 CrossRef CAS.
- H. O. House, J. Org. Chem., 1969, 34, 2324 CrossRef CAS.
- H. Teichmann and V. Prey, Justus Liebigs Ann. Chem., 1970, 732, 121 CrossRef CAS.
- R. Ballini and M. Petrini, Tetrahedron, 2004, 60, 1017 CrossRef CAS PubMed.
- W. E. Noland, Chem. Rev., 1955, 55, 137 CrossRef CAS.
- G. A. Olah and B. G. Gupta, Synthesis, 1980, 44 CrossRef CAS.
- K. Stelion and M. A. Poupart, J. Org. Chem., 1985, 50, 4971 CrossRef.
- R. S. Varma, M. Varma and G. W. Kabalka, Tetrahedron Lett., 1985, 26, 3777 CrossRef CAS.
- M. S. Mourad, R. S. Varma and G. W. Kabalka, Synthesis, 1985, 654 CrossRef CAS PubMed.
- P. S. Vankar, R. Rathore and S. Chandrasekaran, Synth. Commun., 1987, 11, 195 CrossRef.
- S. J. Pennanen, Tetrahedron Lett., 1980, 657 CrossRef CAS.
- J. M. Aizpurua, M. Oiarbide and C. Palomo, Tetrahedron Lett., 1987, 28, 5361 CrossRef CAS.
- C. Palomo, J. M. Aizpurua, F. P. CossIo, J. M. Garcia, M. C. Lopez and M. Oiarbide, J. Org. Chem., 1990, 55, 2070 CrossRef CAS.
- D. Y. Kim and D. Y. Oh, Bull. Korean Chem. Soc., 1997, 18, 994 CAS.
- A. W. Hofmann, Ber. Dtsch. Chem. Ges., 1881, 14, 2725 CrossRef.
- E. S. Wallis and J. F. Lane, Org. React., 1946, 3, 267 Search PubMed.
- T. Shioiri, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 6, p. 795 Search PubMed.
- K. Miyamoto, N. Tada and M. Ochiai, J. Am. Chem. Soc., 2007, 129, 2772 CrossRef CAS PubMed.
- M. Ochiai, K. Miyamoto, Y. Yokota, T. Suefuji and M. Shiro, Angew. Chem., Int. Ed., 2005, 44, 75 CrossRef CAS PubMed.
- K. Miyamoto, Y. Sakai, S. Goda and M. Ochiai, Chem. Commun., 2012, 48, 982 RSC.
- H. D. Dakin, H. T. Clarke and E. R. Taylor, Catechol. Org. Synth., Coll, 1941, 1, 149 Search PubMed.
- N. H. Nam, Y. Kim, Y. J. You, D. H. Hong, H. M. Kim and B. Z. Ahn, Bioorg. Med. Chem. Lett., 2002, 12, 2345 CrossRef CAS.
- I. M. Godfrey, M. V. Sargent and J. A. Elix, J. Chem. Soc., Perkin Trans. 1, 1974, 1353 RSC.
- M. Matsumoto, H. Kobayashi and Y. Hotta, J. Org. Chem., 1984, 49, 4740 CrossRef CAS.
- L. Syper, Synthesis, 1989, 3, 167 CrossRef.
- A. McKillop and W. R. Sanderson, Tetrahedron, 1995, 51, 6145 CrossRef CAS.
- A. Roy, K. R. Reddy, P. K. Mohanta, H. Ila and H. Junjappa, Synth. Commun., 1999, 29, 3781 CrossRef CAS.
- R. S. Varma and K. P. Naicker, Org. Lett., 1999, 1, 189 CrossRef CAS.
- J. L. Zambrano and R. Dorta, Synlett, 2003, 1545 CrossRef CAS.
- E. T. Silva, C. A. Camara, O. A. C. Antunes, E. J. Barreiro and C. A. M. Fraga, Synth. Commun., 2008, 38, 784 CrossRef.
- R. G. F. Giles, I. R. Green and N. Eeden, Synth. Commun., 2006, 36, 1695 CrossRef CAS.
- R. A. Limaye, P. Gaur, M. V. Paradkar and A. D. Natu, Synth. Commun., 2012, 42, 313 CrossRef CAS.
- D. Magdziak, S. J. Meek and T. R. R. Pettus, Chem. Rev., 2004, 104, 1383 CrossRef CAS PubMed.
- S. Rodriguez and P. Wipf, Synthesis, 2004, 2767 CAS.
- R. M. Moriarty and O. Prakash, Org. React., 2001, 57, 327 CAS.
- M. A. Ciufolini, N. A. Braun, S. Canesi, M. Ousmer, J. Chang and D. Chai, Synthesis, 2007, 3759 CrossRef CAS PubMed.
- S. Quideau, L. Pouysegu and D. Deffieux, Synlett, 2008, 467 CrossRef CAS PubMed.
- R. C. Cambie, B. G. Lindsay, P. S. Rutledge and P. D. Woodgate, J. Chem. Soc., Chem. Commun., 1978, 919 RSC.
- H. J. Reich and S. L. Peake, J. Am. Chem. Soc., 1978, 100, 4888 CrossRef CAS.
- R. I. Davidson and P. J. Kropp, J. Org. Chem., 1982, 47, 1904 CrossRef CAS.
- S. Yamamoto, H. Itani, T. Tsuji and W. Nagata, J. Am. Chem. Soc., 1983, 105, 2908 CrossRef CAS.
- N. S. Zefirov, V. V. Zhdankin, G. V. Makhon'kova, Y. V. Dan'kov and A. S. Koz'min, J. Org. Chem., 1985, 50, 187 Search PubMed.
- D. G. Morris and A. G. Shepherd, J. Chem. Soc., Chem. Commun., 1981, 1250 RSC.
- R. A. Moss, K. Bracken and T. J. Emge, J. Org. Chem., 1995, 60, 7739 CrossRef CAS.
- H. Tohma, A. Maruyama, A. Maeda, T. Maegawa, T. Dohi, M. Shiro, T. Morita and Y. Kita, Angew. Chem., Int. Ed., 2004, 43, 3595 CrossRef CAS PubMed.
- T. Dohi, A. Maruyama, N. Takenaga, K. Senami, Y. Minamitsuji, H. Fujioka, S. B. Caemmerer and Y. Kita, Angew. Chem., Int. Ed., 2008, 47, 3787 CrossRef CAS PubMed.
- T. Dohi, Y. Kita and A. Maruyama, Jpn. Kokai Tokkyo Koho 2009149564A, 2009.
- M. Uyanik, T. Yasui and K. Ishihara, Angew. Chem., Int. Ed., 2010, 49, 2175 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Tetrahedron, 2010, 66, 5841 CrossRef CAS PubMed.
- C. Molte-Leth and K. A. Jorgenson, Acta Chem. Scand., 1993, 47, 1117 CrossRef PubMed.
- D. W. Visserr, in Synthetic Procedures in Nucleic Acid Chemistry, ed. W. W. Zorbach and R. S. Tipson, Wiley, New York, 1968, vol. 1, p. 409 Search PubMed.
- J. Duval and J. P. Ebel, Bull. Soc. Chim. Belg., 1964, 46, 1059 CAS.
- T. K. Fukuhara and D. W. Visser, J. Biol. Chem., 1951, 190, 95 CAS.
- R. E. Beltz and D. W. Visser, J. Am. Chem. Soc., 1955, 77, 736 CrossRef CAS.
- A. M. Michelson, J. Am. Chem. Soc., 1958, 80, 1967 CrossRef.
- T. K. Fukuhara and D. W. Visser, J. Am. Chem. Soc., 1959, 81, 1756 CrossRef.
- A. Matauka, H. Inoue and T. Ueda, Chem. Pharm. Bull., 1978, 26, 2340 CrossRef.
- T. L. V. Ulbright, Purines, pyrimidines and Nucleotides, Pergamon, London, 1964 Search PubMed.
- J. B. Hobbs, in Comprehensive Medicinal Chemistry, C. Hansch, P. G. Sammes and J. B. Taylor, Pergamon, Oxford 1990, vol. 2, p. 299 Search PubMed.
- E. K. Ryu and M. MacCoss, J. Org. Chem., 1981, 46, 2819 CrossRef CAS.
- N. Zanatta, L. Fantinel, L. S. Fernandes, A. D. Wouters, H. G. Bonacorso and M. A. P. Martins, Synthesis, 2008, 3492 CrossRef CAS PubMed.
- C. H. Hwang, J. S. Park, J. H. Won, J. N. Kim and E. K. Ryu, Arch. Pharmacal Res., 1992, 15, 69 CrossRef CAS.
- D. A. R. Happer and J. Vaughan, in The Chemistry of the Hydroxyl Group, ed. S. Patai, Interscience Publishers, New York, 1971, Part 1, p. 418 Search PubMed.
- R. Stroh, in Methoden der Organischen Chemie (Houben-Weyl), ed. E. Muller, Georg Thieme Verlag Stuttgart, Stuttgart, 1962, Bd. V/3. S., p. 679 Search PubMed.
- R. H. Huston and A. H. Neeley, J. Am. Chem. Soc., 1935, 57, 2176 CrossRef CAS.
- D. R. Harvey and R. O. C. Norman, J. Chem. Soc., 1961, 3604 RSC.
- D. Ginsburg, J. Am. Chem. Soc., 1951, 73, 2723 CrossRef.
- D. Masilamani and M. M. Rogic, J. Org. Chem., 1981, 46, 4486 CrossRef CAS.
- M. Stebnik, R. Mechoulam and I. Yona, J. Chem. Soc., Perkin Trans. 1, 1987, 1423 RSC.
- E. M. Kosower, W. J. Cole, G. S. Wu, D. E. Cardy and G. Meisters, J. Org. Chem., 1963, 28, 630 CrossRef CAS.
- H. Lubbecke and P. Bolt, Tetrahedron, 1978, 34, 1577 CrossRef.
- D. Bogaext-Vehoogen and R. H. Martin, Bull. Soc. Chim. Belg., 1949, 58, 567 CrossRef.
- K. Ziegler, A. Spath, E. Schaaf, W. Schumann and E. Winkelmann, Ann. Chim., 1942, 551, 82 Search PubMed.
- K. H. Chung, H. J. Kim, H. R. Kim and E. K. Ryu, Synth. Commun., 1990, 20, 2991 CrossRef CAS.
- W. W. Reed and K. J. P. Orton, J. Chem. Soc., 1907, 1553 Search PubMed.
- E. C. Kleiderer and R. Adams, J. Am. Chem. Soc., 1933, 55, 4219 CrossRef CAS.
- C. Musante and P. Fuso, Gazz. Chim. Ital., 1936, 66, 639 CAS.
- C. Beard and W. J. Hickinbottom, J. Chem. Soc., 1958, 2982 RSC.
- L. F. Fieser and D. M. Bowen, J. Am. Chem. Soc., 1940, 62, 2106 Search PubMed.
- G. K. Chip and J. S. Grossert, Can. J. Chem., 1972, 50, 1233 CrossRef CAS.
- S. Kajigaeshi, Y. Shinmasu, S. Fujisaki and T. Kakinami, Bull. Chem. Soc. Jpn., 1990, 63, 941 CrossRef CAS.
- K. H. Chung, K. M. Kim, J. N. Kim and E. K. Ryu, Synth. Commun., 1991, 21, 1917 CrossRef CAS.
- V. Verhe and N. De Kimpe, The Chemistry of Functional Groups-Supplement D, The Chemistry of Halides, Pseudohalides, and Azides, Part 1, ed. S. Patai and Z. Rappoport, J. Wiley and Sons, New York, 1983, ch. 19 Search PubMed.
- R. H. Levin, B. J. Magerlein, A. V. McIntosh Jr, A. R. Hanze, G. S. Fonken, J. L. Thompsom, A. M. Searcy, M. A. Scheri and E. S. Gutsbell, J. Am. Chem. Soc., 1953, 75, 502 CrossRef CAS.
- F. Mukawa, J. Chem. Soc. Jpn., 1957, 78, 450 CrossRef CAS.
- J. Otto and K. Paluch, Rocz. Chem., 1973, 47, 967 (Chem. Abstr., 1973, 79, 136679s) CAS.
- T. Yamauchi, K. Hattori, S. Mizutaki, K. Tamak and S. Uemura, Bull. Chem. Soc. Jpn., 1986, 59, 3617 CrossRef CAS.
- M. A. Kim, H. R. Kim and E. K. Ryu, Synth. Commun., 1990, 20, 1625 CrossRef.
- K. M. Kim, K. H. Chung, J. N. Kim and E. K. Ryu, Synthesis, 1993, 283 CrossRef CAS PubMed.
- H. J. Kim, H. R. Kim, J. N. Kim and E. K. Ryu, Bull. Korean Chem. Soc., 1990, 11, 184 CAS.
- N. Inukai, H. Iwamoto, T. Tamura, I. Yanagisawa, Y. Ishii and M. Murakami, Chem. Pharm. Bull., 1976, 24, 820 CrossRef CAS.
- K. K. Kim, J. N. Kim, K. M. Kim, H. R. Kim and E. K. Ryu, Chem. Lett., 1992, 603 CrossRef CAS.
- Y. Suzuki, Y. Ishiwata, K. Moriyama and H. Togo, Tetrahedron Lett., 2010, 51, 5950 CrossRef CAS PubMed.
- R. D. Bach, C. Canepa, J. E. Winter and P. E. Blanchette, J. Org. Chem., 1997, 62, 5191 CrossRef CAS.
- J. O. Edwards, Peroxide Reaction Mechanisms, ed. J. O. Edwards, Interscience, New York, 1962, p. 67 Search PubMed.
- B. M. Lynch and K. H. Pausacker, J. Chem. Soc., 1955, 1525 RSC.
- N. N. Schwartz and J. H. Blumbergs, J. Org. Chem., 1964, 29, 1976 CrossRef CAS.
- M. Vilka, Bull. Soc. Chim. Fr., 1959, 1401 Search PubMed.
- P. Renolen and J. Ugelstad, J. Chem. Phys., 1960, 57, 634 CAS.
- H. O. House and R. S. Ro, J. Am. Chem. Soc., 1958, 80, 2428 CrossRef CAS.
- Y. Ogata and I. Tabushi, J. Am. Chem. Soc., 1961, 83, 3440 CrossRef CAS.
- R. Curci, R. A. Di Prete, J. O. Edwards and G. Modena, J. Org. Chem., 1970, 30, 740 CrossRef.
- P. D. Bartlett, Rec. Chem. Prog., 1950, 11, 47 CAS.
- R. D. Bach, C. L. Willis and J. M. Domagals, Applications of MO Theory in Organic Chemistry, I. C. Csizmadia, Elsevier Scientific, Amsterdam, 1977, vol. 2, p. 221 Search PubMed.
- R. D. Bach, J. L. Andres and F. Davis, J. Org. Chem., 1992, 57, 613 CrossRef CAS.
- M. J. Crossley and A. W. Stamford, Aust. J. Chem., 1993, 46, 1443 CrossRef CAS.
- G. W. Holbert, B. Ganem, D. Van Engen, J. Clardy, L. Borsub, K. Chantrapromma, C. Sadavongvivad and Y. Thebtaranonth, Tetrahedron Lett., 1979, 20, 715 CrossRef.
- M. Souchet, M. Baillarge and F. Le Goffic, Tetrahedron Lett., 1988, 29, 191 CrossRef CAS.
- H. Wild, J. Org. Chem., 1994, 59, 2748 CrossRef CAS.
- S. Takano, M. Moriya and K. Ogasawara, J. Chem. Soc., Chem. Commun., 1993, 614 RSC.
- H. Shimizu, H. Okamura, T. Iwagawa and M. Nakatani, Tetrahedron Lett., 2001, 57, 1903 CrossRef CAS.
- A. B. Smith and R. E. Richmond, J. Org. Chem., 1981, 46, 4814 CrossRef CAS.
- A. B. Smith and R. E. Richmond, J. Am. Chem. Soc., 1983, 105, 575 CrossRef CAS.
- R. Baker, C. L. Gibson, C. J. Swain and D. J. Tapolczay, J. Chem. Soc., Chem. Commun., 1984, 619 RSC.
- R. Baker, C. L. Gibson, C. J. Swain and D. J. Tapolczay, J. Chem. Soc., Perkin Trans. 1, 1985, 1509 RSC.
- I. Yamamoto and K. Narasaka, Bull. Chem. Soc. Jpn., 1994, 67, 3327 CrossRef CAS.
- S. Ogawa and T. Takagaki, J. Org. Chem., 1985, 50, 2356 CrossRef CAS.
- V. Lorbach, D. Franke, M. Nieger and M. Muller, Chem. Commun., 2002, 494 RSC.
- C. Li, E. A. Pace, M. C. Liang, E. Lobkovsky, T. D. Gilmore and J. A. Porco, J. Am. Chem. Soc., 2001, 123, 11308 CrossRef CAS.
- O. Block, G. Klein, H. J. Altenbach and D. J. Brauer, J. Org. Chem., 2000, 65, 716 CrossRef CAS PubMed.
- S. T. Murphy, J. R. Bencsik and C. R. Johnson, Org. Lett., 1999, 1, 1483 CrossRef CAS.
- J. Marco-Contelles, M. T. Molina and S. Anjum, Chem. Rev., 2004, 104, 2857 CrossRef CAS PubMed.
- A. G. Martınez, E. T. Vilar, A. G. Fraile, S. M. Cerero, P. M. Ruiz, C. D. Morillo and B. L. Maroto, Tetrahedron Lett., 2007, 48, 5981 CrossRef PubMed.
- T. L. Shih and Y. L. Lin, Synth. Commun., 2005, 35, 1809 CrossRef CAS PubMed.
- C. Rogers, Y. Shen, D. Burgoyne and E. Piers, Synth. Commun., 2002, 32, 2991 CrossRef CAS PubMed.
- J. Benites, M. D. Preite and M. Cortes, Synth. Commun., 2001, 31, 1347 CrossRef CAS PubMed.
- E. Lassabaa, H. El Jamilia, A. Chekrouna, A. Benharrefa, A. Chiaronib, C. Richeb and J. P. Lavergne, Synth. Commun., 1998, 28, 2641 CrossRef.
- D. M. Gleavet, B. M. Savalls and W. W. McWhorte, Synth. Commun., 1997, 27, 2425 CrossRef.
- G. Moyna, H. J. Williams and A. I. Scott, Synth. Commun., 1996, 26, 2235 CrossRef CAS.
- O. Muraoka, Y. Wang, M. Okumura and S. Nishiura, Synth. Commun., 1996, 26, 1555 CrossRef CAS.
- K. T. Kang and J. Sun, Synth. Commun., 1995, 25, 2647 CrossRef CAS.
- K. T. Kang, J. Sun, S. S. Hwang and K. K. Jyung, Synth. Commun., 1994, 24, 2915 CrossRef CAS.
- C. Y. Huang, C. Y. Cabell and E. V. Anslyn, Synth. Commun., 1994, 24, 2757 CrossRef CAS.
- M. T. Le Goff, C. Marazano, J. L. Fourrey and B. C. Das, Synth. Commun., 1982, 12, 891 CrossRef CAS.
- F. Benfatti, G. Cardillo, L. Gentilucci, R. Perciaccante and A. Tolomelli, Synlett, 2005, 2204 CAS.
- Z. Song, X. Su, K. Huang, H. Lin, F. Liu and J. Tang, Polymer, 2011, 52, 4456 CrossRef CAS PubMed.
- N. K. Jana and J. G. Verkade, Org. Lett., 2003, 5, 3787 CrossRef CAS PubMed.
- W. Adam, A. G. Griesbeck and X. Wang, Liebigs Ann. Chem., 1992, 193 CrossRef CAS.
- F. Camps, J. Coll, A. Messeguer and F. Pujol, J. Org. Chem., 1982, 47, 5402 CrossRef CAS.
- M. Freccero, R. Gandolfi, M. Sarzi-Amade and A. Rastelli, J. Org. Chem., 2005, 70, 9573 CrossRef CAS PubMed.
- C. Kim, T. G. Traylor and C. L. Perrin, J. Am. Chem. Soc., 1998, 120, 9513 CrossRef CAS.
- A. K. Feldman, B. Colasson, K. B. Sharpless and V. V. Fokin, J. Am. Chem. Soc., 2005, 127, 13444 CrossRef CAS PubMed.
- Y. S. Angelis and M. Orfanopoulos, J. Org. Chem., 1997, 62, 6083 CrossRef CAS.
- R. S. Iyer and T. M. Harris, Chem. Res. Toxicol., 1993, 6, 313 CrossRef CAS.
- Z. Solati, M. Hashemi and L. Ebrahimi, Catal. Lett., 2011, 141, 163 CrossRef CAS PubMed.
- J. D. Ha, E. Y. Shin, Y. Chung and J. K. Choi, Bull. Korean Chem. Soc., 2003, 24, 1567 CrossRef CAS.
- K. Kishikawa, M. Naruse, S. Kohmoto, M. Yamamoto and K. Yamaguchi, J. Chem. Soc., Perkin Trans. 1, 2001, 462 RSC.
- J. D. Butler, M. B. Donald, Z. Ding, J. C. Fettinger and M. J. Kurth, Tetrahedron Lett., 2009, 50, 5110 CrossRef CAS PubMed.
- F. Fringguelli, R. Germani, F. Pizzo and G. Savelli, Tetrahedron Lett., 1989, 30, 1427 CrossRef.
- A. Kumar and V. Bhakuni, Tetrahedron Lett., 1996, 37, 4751 CrossRef CAS.
- T. Ono and P. Henderson, Tetrahedron Lett., 2002, 43, 7961 CrossRef CAS.
- A. Chekroun, A. Jarid, A. Benharref and A. Boutalib, J. Mol. Struct.: THEOCHEM, 2002, 588, 201 CrossRef CAS.
- S. L. Schreiber, T. Sammakia, B. Huh and G. Schulte, J. Am. Chem. Soc., 1986, 108, 2106 CrossRef CAS.
- A. S. Rao, Addition reactions with formation of Carbon-Oxygen bonds, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 7, p. 357 Search PubMed.
- W. Adam, S. G. Bosio and B. T. Wolff, Org. Lett., 2003, 5, 819 CrossRef CAS PubMed.
- C. W. Bond, A. J. Cresswell, S. G. Davies, A. M. Fletcher, W. Kurosawa, J. A. Lee, P. M. Roberts, A. J. Russell, A. D. Smith and J. E. Thomson, J. Org. Chem., 2009, 74, 6735 CrossRef CAS PubMed.
- M. S. F. L. K. Jie and C. K. Lam, Ultrason. Sonochem., 1995, 2, S11 CrossRef.
- P. Fristrup, P. R. Lassen, D. Tanner and K. J. Jalkanen, Theor. Chem. Acc., 2008, 119, 133 CrossRef CAS PubMed.
- B. Gopishetty, S. Gogoi and A. K. Dutta, Tetrahedron: Asymmetry, 2011, 22, 1081 CrossRef CAS PubMed.
- T. Chevtchouk, J. Ollivier and J. Salauen, Tetrahedron: Asymmetry, 1997, 8, 1011 CrossRef CAS.
- E. B. Rodriguez and D. Rodriguez-Amaya, J. Food Sci., 2009, 74, C674 CrossRef CAS PubMed.
- J. K. Ekegren, T. Unge, M. Z. Safa, H. Wallberg, B. Samuelsson and A. Hallberg, J. Med. Chem., 2005, 48, 8098 CrossRef CAS PubMed.
- J. R. Luly, N. Yi, J. Soderquist, H. Stein, J. Cohen, T. J. Perun and J. J. Plattner, J. Med. Chem., 1987, 30, 1609 CrossRef CAS.
- J. R. Luly, G. Bolis, N. BaMaung, J. Soderquist, J. F. Dellaria, H. Stein, J. Cohen, T. J. Perun, J. Greer and J. J. Plattnert, J. Med. Chem., 1988, 31, 532 CrossRef CAS.
- J. Tang, Y. Zu, W. Huo, L. Wang, J. Wang, M. Jia, W. Zhang and W. R. Thiel, J. Mol. Catal. A: Chem., 2012, 355, 201 CrossRef CAS PubMed.
- W. Adam, A. Corma, A. Martinez, C. M. Mitchell, T. I. Reddy, M. Renz and A. K. Smerz, J. Mol. Catal. A: Chem., 1997, 117, 357 CrossRef CAS.
- M. R. Klaas, M. Kunz and S. Warwel, J. Mol. Catal. B: Enzym., 1999, 7, 283 CrossRef.
- M. C. Pirrung and G. M. McGeehan, J. Org. Chem., 1983, 48, 5144 CrossRef.
- R. Jay, J. R. Luly, J. F. Dellaria, J. J. Plattner, J. L. Soderquist and N. Yi, J. Org. Chem., 1987, 52, 1487 CrossRef.
- F. Matthew, M. F. Schlecht and H. Kim, J. Org. Chem., 1989, 54, 583 CrossRef.
- T. Kume and K. Akiba, J. Org. Chem., 1989, 54, 1935 CrossRef CAS.
- D. B. Berkowitz and M. L. Pedersen, J. Org. Chem., 1995, 60, 5368 CrossRef CAS.
- R. Eckrich, B. Neumann, H. G. Stammler and D. Kuck, J. Org. Chem., 1996, 61, 3839 CrossRef CAS PubMed.
- F. Benedetti, S. Miertus, S. Norbedo, A. Tossi and P. Zlatoidzky, J. Org. Chem., 1997, 62, 9348 CrossRef CAS.
- W. Adam, S. G. Bosio, N. J. Turro and B. T. Wolff, J. Org. Chem., 2004, 69, 1704 CrossRef CAS PubMed.
- S. Huang, Z. Xiao, F. Wang, L. Gan, X. Zhang, X. Hu, S. Zhang, M. Lu, Q. Pan and L. Xu, J. Org. Chem., 2004, 69, 2442 CrossRef CAS PubMed.
- V. Cere, F. Peri, S. Pollicino, A. Ricci, F. J. Devlin, P. J. Stephens, F. Gasparrini, R. Rompietti and C. Villani, J. Org. Chem., 2005, 70, 664 CrossRef CAS PubMed.
- K. D. Safa, K. Ghorbanpour, A. Hassanpour and S. Tofangdarzadeh, J. Organomet. Chem., 2009, 694, 1907 CrossRef CAS PubMed.
- D. T. Williamson, B. D. Mather and T. E. Long, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 84 CrossRef CAS.
- F. R. De Risi, L. D. Ilario and A. Martinelli, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3082 CrossRef CAS.
- K. H. Budt, J. M. Vatele and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 6080 CrossRef CAS PubMed.
- T. G. Traylor and F. Xu, J. Am. Chem. Soc., 1988, 110, 1953 CrossRef CAS.
- E. Vedejs and W. H. Dent, J. Am. Chem. Soc., 1989, 111, 6861 CrossRef CAS.
- J. A. Marshall and W. J. DuBay, J. Am. Chem. Soc., 1992, 114, 1450 CrossRef CAS.
- K. J. Shea and J. S. Kim, J. Am. Chem. Soc., 1992, 114, 3044 CrossRef CAS.
- W. Namt and J. S. Valentine, J. Am. Chem. Soc., 1993, 115, 1772 CrossRef.
- D. A. Singleton, S. R. Merrigan, J. Liu and K. N. Houk, J. Am. Chem. Soc., 1997, 119, 3385 CrossRef CAS.
- X. Chen, Z. Su, J. H. Horner and M. Newcomb, Org. Biomol. Chem., 2011, 9, 7427 CAS.
- G. Mehta, S. R. Singh, P. Balanarayan and S. R. Gadre, Org. Lett., 2002, 4, 2297 CrossRef CAS PubMed.
- Q. Zhu, M. Uttamchandani, D. Li, M. L. Lesaicherre and S. Q. Yao, Org. Lett., 2003, 5, 1257 CrossRef CAS PubMed.
- W. Adam, A. Pastor, K. Peters and E. M. Peters, Org. Lett., 2000, 2, 1019 CrossRef CAS.
- J. S. Prasad, T. Vu, M. J. Totleben, G. A. Crispino, D. J. Kacsur, S. Swaminathan, J. E. Thornton, A. Fritz and A. K. Singh, Org. Process Res. Dev., 2003, 7, 821 CrossRef CAS.
- R. Alonso, Pharm. Chem. J., 2006, 40, 476 CrossRef CAS PubMed.
- X. Zhang, P. Geoffroy, M. Miesch, D. Julien-David, F. Raul, D. Aoude-Werner and E. Marchioni, Steroids, 2005, 70, 886 CrossRef CAS PubMed.
- M. Ahmar, S. Thomé and B. Cazes, Synlett, 2006, 2, 279 Search PubMed.
- E. L. Williams, Synth. Commun., 1992, 22, 1017 CrossRef CAS.
- V. I. Hugo and T. Masenya, Synth. Commun., 2006, 36, 1631 CrossRef CAS.
- D. Frank, S. I. Kozhushkov, T. Labahn and A. de Meijere, Tetrahedron, 2002, 58, 7001 CrossRef CAS.
- K. Uchida, K. Ishigami, H. Watanabe and T. Kitahara, Tetrahedron, 2007, 63, 1281 CrossRef CAS PubMed.
- Z. Fang, G. E. Agoston, G. Ladouceur, A. M. Treston, L. Wang and M. Cushman, Tetrahedron, 2009, 65, 10535 CrossRef CAS PubMed.
- K. Krohn and J. Micheel, Tetrahedron, 1998, 54, 4827 CrossRef CAS.
- A. P. Marchand, B. Ganguly, R. Shukla, K. Krishnudu and V. S. Kumar, Tetrahedron, 1999, 55, 8313 CrossRef CAS.
- M. Freccero, R. Gandolfi and M. Sarzi-Amade, Tetrahedron, 1999, 55, 11309 CrossRef CAS.
- A. Garcia-Granados, P. E. Lopez, E. Melguizo, A. Parra and Y. Simeo, Tetrahedron, 2004, 60, 3831 CrossRef CAS PubMed.
- S. Romeo and D. H. Rich, Tetrahedron Lett., 1993, 34, 7187 CrossRef CAS.
- K. Ohmori, S. Nishiyama and S. Yamamura, Tetrahedron Lett., 1995, 36, 6519 CrossRef CAS.
- H. J. Mitchell and S. Warren, Tetrahedron Lett., 1996, 37, 2105 CrossRef CAS.
- F. Minutolo, D. Pini and P. Salvadori, Tetrahedron Lett., 1996, 37, 3375 CrossRef CAS.
- K. Maruyama, M. Ueda, S. Sasaki, Y. Iwata, M. Miyazawa and M. Miyashita, Tetrahedron Lett., 1998, 39, 4517 CrossRef CAS.
- A. P. Marchand, S. Alihodzic and E. Z. Dong, Tetrahedron Lett., 1998, 39, 8055 CrossRef CAS.
- S. E. de Sousa, P. O'Brien, C. D. Pilgram, D. Roder and T. D. Towers, Tetrahedron Lett., 1999, 40, 391 CrossRef CAS.
- S. Saito, H. Itoh, Y. Ono, K. Niihioka and T. Moriwake, Tetrahedron: Asymmetry, 1993, 4, 5 CrossRef CAS.
- F. Minutolo, D. Pini, A. Petri and P. Salvadori, Tetrahedron: Asymmetry, 1996, 7, 2293 CrossRef CAS.
- Y. Iwata, N. Maekawara, K. Tanino and M. Miyashita, Angew. Chem., Int. Ed., 2005, 44, 1532 CrossRef CAS PubMed.
- M. A. González, L. Agudelo and L. Betancur-Galvis, Antiviral Res., 2010, 85, 562 CrossRef PubMed.
- M. A. Brimble, C. J. Funnell, S. Gorsuch and M. Sidford, ARKIVOC, 2001, 74 CrossRef CAS.
- I. Jastrzębska, K. S. Katryński and J. W. Morzycki, ARKIVOC, 2002, 46 CrossRef.
- M. N. Bin Omar, R. J. Hamilton and H. A. Moynihan, ARKIVOC, 2003, 190 CrossRef CAS.
- D. A. Alonso, A. Babrowski, M. Fuensanta, C. Nájera and M. Varea, ARKIVOC, 2007, 243 CrossRef CAS.
- T. A. Hill, S. G. Stewart, S. P. Ackland, J. Gilbert, B. Sauer, J. A. Sakoff and A. McCluskey, Bioorg. Med. Chem., 2007, 15, 6126 CrossRef CAS PubMed.
- I. Hueso-Falcón, N. Girón, P. Velasco, J. M. Amaro-Luis, A. G. Ravelo, B. de las Heras, S. W. Hortelano and A. Estevez-Braun, Bioorg. Med. Chem., 2010, 18, 1724 CrossRef PubMed.
- Q. Zhang, Z. Y. Jiang, J. Luo, P. Cheng, Y. B. Ma, X. M. Zhang, F. X. Zhang, J. Zhou and J. J. Chen, Bioorg. Med. Chem. Lett., 2008, 18, 4647 CrossRef CAS PubMed.
- C. Marquissolo, A. de Fátima, L. K. Kohn, A. L. T. G. Ruiz, J. E. de Carvalho and R. A. Pilli, Bioorg. Chem., 2009, 37, 52 CrossRef CAS PubMed.
- N. Kutsumura and S. Nishiyama, Bull. Chem. Soc. Jpn., 2006, 79, 468 CrossRef CAS.
- J. Kwak, J. K. In, M. S. Lee, J. Y. Yu, Y. P. Yun, J. T. Hong, S. J. Lee, S. Y. Seo, S. K. Ahn, Y. G. Suh, B. Y. Hwang, H. Lee, K. H. Min and J. Y. Jung, Bull. Korean Chem. Soc., 2009, 30, 2881 CrossRef CAS.
- P. E. Georghiou and Y. Ren, Can. J. Chem., 1993, 71, 364 CrossRef CAS.
- J. G. Kim, D. W. Park and Y. S. Tak, Catal. Lett., 2000, 65, 127 CrossRef.
- M. B. Andrus and B. W. Poehlein, Tetrahedron Lett., 2000, 41, 1013 CrossRef CAS.
- T. Nishinaga, T. Uto, R. Inoue, A. Matsuura, N. Treitel, M. Rabinovitz and K. Komatsu, Chem.–Eur. J., 2008, 14, 2067 CrossRef CAS PubMed.
- S. H. Lee, L. Xu, B. K. Park, Y. V. Mironov, S. H. Kim, Y. J. Song, C. Kim, Y. Kim and S. J. Kim, Chem.–Eur. J., 2010, 16, 4678 CrossRef CAS PubMed.
- Y. J. Song, S. H. Lee, H. M. Park, S. H. Kim, H. G. Goo, G. H. Eom, J. H. Lee, M. S. Lah, Y. Kim, S. J. Kim, J. E. Lee, H. I. Lee and C. Kim, Chem.–Eur. J., 2011, 17, 7336 CrossRef CAS PubMed.
- Y. J. Song, M. Y. Hyun, J. H. Lee, H. G. Lee, J. H. Kim, S. P. Jang, J. Y. Noh, Y. K. Kim, S. J. Kim, S. J. Lee and C. Kim, Chem.–Eur. J., 2012, 18, 6094 CrossRef CAS PubMed.
- A. Dermenci, P. S. Selig, R. A. Domaoal, K. A. Spasov, K. S. Anderson and S. Miller, Chem. Sci., 2011, 2, 1568 RSC.
- A. de Meijere, H. Wenck, S. Zöllner, P. Merstetter, A. Arnold, F. Gerson, P. R. Schreiner, R. Boese, D. Bläser, R. Gleiter and S. I. Kozhushkov, Chem.–Eur. J., 2001, 7, 5382 CrossRef CAS.
- M. Kuba, N. Furuichi and S. Katsumura, Chem. Lett., 2002, 12, 1248 CrossRef.
- M. Bols, R. G. Hazell and I. B. Thomsen, Chem.–Eur. J., 1997, 3, 940 CrossRef CAS.
- T. Linker, F. Rebien, G. Toth, A. Simon, J. Kraus and G. Bringmann, Chem.–Eur. J., 1998, 4, 1944 CrossRef CAS.
- Z. Shu-Jia, C. Yong-Kang, L. Hui-Ming, H. Wei-Yang, R. Viktor and M. Peter, Chin. J. Chem., 2006, 24, 681 CrossRef.
- A. V. Moro, M. R. dos Santos and C. R. D. Correia, Eur. J. Org. Chem., 2011, 7259 CrossRef CAS.
- M. Ahmar, S. Thomé and B. Cazes, Eur. J. Org. Chem., 2012, 7093 CrossRef CAS.
- W. Adam, C. M. Mitchell and C. R. Saha-Möller, Eur. J. Org. Chem., 1999, 785 CrossRef CAS.
- M. Yamamoto, H. Yamazawa, N. Nakajima and T. Ando, Eur. J. Org. Chem., 1999, 1503 CrossRef CAS.
- I. Bombarda, L. Cezanne and E. M. Gaydou, Flavour Fragrance J., 2004, 19, 275 CrossRef CAS.
- K. D. Safa and A. Hassanpour, J. Polym. Res., 2010, 17, 719 CrossRef CAS PubMed.
- G. A. O'Doherty, R. D. Rogers and L. A. Paquette, J. Am. Chem. Soc., 1994, 116, 10883 CrossRef CAS.
- E. Vedejs, W. H. Dent, J. T. Kendall and P. A. Oliver, J. Am. Chem. Soc., 1996, 118, 3556 CrossRef CAS.
- W. Nam, M. H. Lim, H. J. Lee and C. Kim, J. Am. Chem. Soc., 2000, 122, 6641 CrossRef CAS.
- G. A. Molander and M. Ribagorda, J. Am. Chem. Soc., 2003, 125, 11148 CrossRef CAS PubMed.
- A. K. Kelly, A. E. Lurain and P. J. Walsh, J. Am. Chem. Soc., 2005, 127, 14668 CrossRef CAS PubMed.
- S. Wan, H. Gunaydin, K. N. Houk and P. E. Floreancig, J. Am. Chem. Soc., 2007, 129, 7915 CrossRef CAS PubMed.
- C. Ebner, C. A. Müller, C. Markert and A. Pfaltz, J. Am. Chem. Soc., 2011, 133, 4710 CrossRef CAS PubMed.
- M. Mendelovici and E. Glotter, J. Chem. Soc., Perkin Trans. 1, 1992, 1735 RSC.
- T. J. Chow, Y. S. Hon, C. C. Jen, S. S. Liu, J. H. Chern and K. J. Lin, J. Chem. Soc., Perkin Trans. 1, 1998, 1095 RSC.
- S. A. Brunton and K. Jones, J. Chem. Soc., Perkin Trans. 1, 2000, 763 RSC.
- H. H. Jensen and M. Bols, J. Chem. Soc., Perkin Trans. 1, 2001, 1, 905 RSC.
- D. Nicoletti, A. A. Ghini, R. F. Baggio, M. T. Garland and G. Burton, J. Chem. Soc., Perkin Trans. 1, 2001, 1511 RSC.
- J. Hudec, J. Huke and J. W. Liebeschuetz, J. Chem. Soc., Perkin Trans. 2, 1998, 1129 RSC.
- R. J. Capon, C. Skene, E. H. T. Liu, E. Lacey, J. H. Gill, K. Heiland and T. Friedel, J. Nat. Prod., 2004, 67, 1277 CrossRef CAS PubMed.
- T. Koerner, H. Slebocka-Tilk and R. S. Brown, J. Org. Chem., 1999, 64, 196 CrossRef CAS PubMed.
- Y. Tajima and K. Takeuchi, J. Org. Chem., 2002, 67, 1696 CrossRef CAS PubMed.
- W. C. Cheng and M. K. Kurth, J. Org. Chem., 2002, 67, 4387 CrossRef CAS PubMed.
- R. K. Pandey, L. Wang, N. J. Wallock, S. Lindeman and W. A. Donaldson, J. Org. Chem., 2008, 73, 7236 CrossRef CAS PubMed.
- K. C. Guérard, A. Guérinot, C. Bouchard-Aubin, M. A. Ménard, M. Lepage, M. A. Beaulieu and S. Canesi, J. Org. Chem., 2012, 77, 2121 CrossRef PubMed.
- M. B. Brennan, T. D. Claridge, R. G. Compton, S. G. Davies, A. M. Fletcher, M. C. Henstridge, D. S. Hewings, W. Kurosawa, J. A. Lee, P. M. Roberts, A. K. Schoonen and J. E. Thomson, J. Org. Chem., 2012, 77, 7241 CrossRef CAS PubMed.
- S. Naito, A. Saito, Y. Furukawa, T. Hata, Y. Nakada, S. Muramatsu and J. Ide, J. Antibiot., 1994, 47, 812 CrossRef CAS.
- R. Tsang, B. Fraser-reid and A. T. McPhail, J. Carbohydr. Chem., 1986, 5, 513 CrossRef CAS.
- R. S. Centko and R. S. Mohan, J. Chem. Educ., 2001, 78, 77 CrossRef CAS.
- M. Bartok and K. L. Lang, The Chemistry of ethers, crown ethers, hydroxyl groups and their Sulfur analogues, ed. S. Patai, The Chemistry of Functional Groups-Part 2 Supplement E 609, 1980 Search PubMed.
- D. Swern, G. N. Bil1en and J. T. Scanland, J. Am. Chem. Soc., 1946, 68, 1504 CrossRef CAS.
- L. N. Owen and P. N. Smith, J. Chem. Soc., 1952, 4026 RSC.
- R. Lombard and G. Schrceder, Bull. Soc. Chim. Fr., 1963, 2800 CAS.
- C. V. Wilson, Org. React., 1957, 9, 332 Search PubMed.
- C. B. Anderson and S. Winstein, J. Org. Chem., 1963, 28, 605 CrossRef CAS.
- A. S. Rao, S. K. Paknikar and J. G. Kirtane, Tetrahedron, 1983, 2323 CrossRef CAS.
- F. Fringuelli, R. Germani, F. Pizzo and G. Savel1i, Synth. Commun., 1989, 19, 1939 CrossRef CAS.
- C. Aciro, T. D. W. Claridge, S. G. Davies, P. M. Roberts, A. J. Russell and J. E. Thomson, Org. Biomol. Chem., 2008, 6, 3751 CAS.
- A. Wu, A. Dehestani, R. Hayoun, W. Kaminsky and J. M. Mayer, Inorg. Chim. Acta, 2009, 362, 4534 CrossRef PubMed.
- Y. B. Kang and L. H. Gade, J. Org. Chem., 2012, 77, 1610 CrossRef CAS PubMed.
- W. Zhong, S. Liu, J. Yang, X. Meng and Z. Li, Org. Lett., 2012, 14, 3336 CrossRef CAS PubMed.
- Y. B. Kang and L. H. Gade, J. Am. Chem. Soc., 2011, 133, 3658 CrossRef CAS PubMed.
- J. B. Sweeney, Chem. Soc. Rev., 2002, 31, 247 RSC.
- W. McCoull and F. A. Davis, Synthesis, 2000, 1347 CrossRef CAS PubMed.
- P. Mueller and C. Fruit, Chem. Rev., 2003, 103, 2905 CrossRef CAS PubMed.
- R. S. Atkinson, Tetrahedron, 1999, 55, 1519 CrossRef CAS.
- A. Yoshimura, K. R. Middleton, C. Zhu, V. N. Nemykin and V. V. Zhdankin, Angew. Chem., Int. Ed., 2012, 51, 8059 CrossRef CAS PubMed.
- Comprehensive of Organic Synthesis (Oxidation), ed. B. M. Trost, Pergamon, New York, 1991, vol. 7 Search PubMed.
- H. R. Kim, J. H. Jung, J. N. Kim and E. K. Ryu, Synth. Commun., 1990, 20, 637 CrossRef CAS.
- S. D. Rychnovsky and R. Vaidyanathan, J. Org. Chem., 1999, 64, 310 CrossRef CAS.
- B. Ganern, J. Org. Chem., 1975, 40, 1998 CrossRef.
- J. A. Cella, J. A. Kelley and E. F. Kenehan, J. Org. Chem., 1975, 40, 1860 CrossRef CAS.
- C. Zhu, L. Ji, Q. Zhang and Y. Wei, Can. J. Chem., 2010, 88, 362 CrossRef CAS PubMed.
- J. A. Cella and J. P. McGrath, Tetrahedron Lett., 1975, 47, 4115 CrossRef.
- S. V. Ley, Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon, Oxford, 1991, vol. 7 Search PubMed.
- S. Kamijo, Y. Amaoka and M. Inoue, Chem.–Asian J., 2010, 5, 486 CrossRef CAS PubMed.
- H. Schwertfeger, C. Würtele and P. R. Schreiner, Synlett, 2010, 493 CAS.
- K. E. Gilbert and W. T. Borden, J. Org. Chem., 1979, 44, 659 CrossRef CAS.
- A. A. Fokin, A. Merz, N. A. Fokina, H. Schwertfeger, S. L. Liu, J. E. P. Dahl, R. M. K. Carlson and P. R. Schreiner, Synthesis, 2009, 909 CrossRef CAS PubMed.
- E. Kun and J. Mendeleyev, Aryl Nitroso Compounds as Specific Inactivators of Retroviral (Asymmetric) Zinc Fingers and as Antitumor Agents, U.S. Patent 5516941, 1996, Chem. Abstr. 1996, 125, 86322.
- B. G. Gowenlock and G. B. Richter-Addo, Chem. Rev., 2004, 104, 3315 CrossRef CAS PubMed.
- S. J. Wratten, H. Fujiwara and R. T. Solsten, J. Agric. Food Chem., 1987, 35, 484 CrossRef CAS.
- E. C. Kimmel, J. E. Casida and L. O. Ruzo, J. Agric. Food Chem., 1986, 34, 157 CrossRef CAS.
- H. H. Baer and S. H. L. Chiu, Can. J. Chem., 1973, 51, 1812 CrossRef CAS.
- J. E. Baldwin, A. K. Qureshi and B. Sklarz, J. Chem. Soc., 1969, 1073 CAS.
- M. Cibian, S. Langis-Barsetti and G. S. Hanan, Synlett, 2011, 405 CAS.
- N. Durust, M. A. Akay, Y. Durust and E. Kilic, Anal. Sci., 2000, 16, 825 CrossRef CAS.
- Y. Dueruest, M. Akcan, O. Martiskainen, E. Siirola and K. Pihlaja, Polyhedron, 2008, 27, 999 CrossRef CAS PubMed.
- R. C. Larock, in Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley-VCH, New York, 2nd edn, 1999 Search PubMed.
- H. M. Meshram, Synth. Commun., 1990, 20, 3253 CrossRef CAS.
- A. I. Bosch, P. De La Cruz, E. Diez-Barra, A. Loupy and F. Langa, Synlett, 1995, 1295 Search PubMed.
- K. V. N. S. Srinivas, E. B. Reddy and B. Das, Synlett, 2002, 625 CrossRef CAS PubMed.
- B. Das, N. Ravindranath, B. Venkataiah and P. Madhusudhan, J. Chem. Res., Synop., 2000, 482 CrossRef CAS.
- B. Das and B. Venkataiah, Synth. Commun., 1999, 29, 863 CrossRef CAS.
- B. Das, P. Madhusudhan and A. Kashinatham, Tetrahedron Lett., 1998, 39, 431 CrossRef CAS.
- B. Das, P. Madhusudhan and A. Kashinatham, Bioorg. Med. Chem. Lett., 1998, 8, 1403 CrossRef CAS.
- A. Lavrent, P. Jacquault, J. L. Di Martino and J. Hamelin, J. Chem. Soc., Chem. Commun., 1995, 1101 RSC.
- F. Delgado, A. C. Cano, O. Garcia, J. Alvarado, L. Velasco, C. Alvarez and H. Rudler, Synth. Commun., 1992, 22, 2125 CrossRef CAS.
- C. Barnett, I. M. Cohn and J. Lincoln, US Pat. 2754298, 1956, Chem. Abstr., 1957, 51, 2853.
- A. Novotny, US Pat. 2569114, 1951, Chem Abstr. 1952, 46, 5078.
- A. Novotny, US Pat. 2579851, 1951, Chem. Abstr., 1952, 46, 6668.
- M. S. Wolf, D. Dutta and J. Aube, J. Org. Chem., 1997, 62, 654 CrossRef PubMed.
- O. Kitagawa, D. Vander Velde, D. Dutta, M. Morton, F. Takusagawa and J. Aube, J. Am. Chem. Soc., 1995, 117, 5169 CrossRef CAS.
- A. J. Post and H. Morrison, J. Am. Chem. Soc., 1995, 117, 7812 CrossRef CAS.
- J. Aube, S. Ghosh and M. Tanol, J. Am. Chem. Soc., 1994, 116, 9009 CrossRef CAS.
- J. Aube and M. S. Wolf, Bioorg. Med. Chem. Lett., 1992, 2, 925 CrossRef CAS.
- J. Aube, Y. Wang, S. Ghosh and K. L. Langhans, Synth. Commun., 1991, 21, 693 CrossRef CAS.
- J. Aube, M. Hammond, E. Gherardini and F. Takusagawa, J. Org. Chem., 1991, 56, 499 CrossRef CAS.
- J. Aube and M. Hammond, Tetrahedron Lett., 1990, 31, 2963 CrossRef CAS.
- J. Aube, Y. Wang, M. Hammond, M. Tanol, F. Takusagawam and D. Vander Velde, J. Am. Chem. Soc., 1990, 112, 4879 CrossRef CAS.
- J. Aube, Tetrahedron Lett., 1988, 29, 4509 CrossRef CAS.
- G. P. Johnson and B. A. J. Marples, J. Chem. Soc., Perkin Trans. 1, 1988, 3399 RSC.
- J. Aube, P. M. Burgett and Y. Wang, Tetrahedron Lett., 1988, 29, 151 CrossRef CAS.
- G. P. Johnson and B. A. Marples, Tetrahedron Lett., 1985, 26, 4115 CrossRef CAS.
- E. Oliveros and M. Riviere et Armand Lattes, J. Heterocycl. Chem., 1980, 17, 1025 CrossRef CAS.
- E. Oliveros, H. Antoun and M. Riviere et Armand Lattes, J. Heterocycl. Chem., 1976, 13, 623 CrossRef CAS.
- S. Y. Kim, G. An and H. Rhee, Synlett, 2003, 112 Search PubMed.
- G. An and H. Rhee, Synlett, 2003, 876 CAS.
- G. An, M. Kim, J. Y. Kim and H. Rhee, Tetrahedron Lett., 2003, 44, 2183 CrossRef CAS.
- J. S. Splitter and M. Calvin, J. Org. Chem., 1965, 30, 3427 CrossRef CAS.
- D. Enders, A. S. Amaya and F. Pierre, New J. Chem., 1999, 23, 261 RSC.
- P. Nongkunsarn and C. A. Ramsden, Tetrahedron, 1997, 53, 3805 CrossRef CAS.
- D. L. Waller, C. R. J. Stephenson and P. Wipf, Org. Biomol. Chem., 2007, 5, 58 CAS.
- E. Wenkert, N. F. Golob, S. S. Sathe and R. A. Smith, Synth. Commun., 1973, 3, 205 CrossRef CAS.
- G. F. Hennion and F. P. Kupiecki, J. Org. Chem., 1953, 18, 1601 CrossRef CAS.
- B. M. Kwon and C. S. Foote, J. Org. Chem., 1989, 54, 3878 CrossRef CAS.
- P. Cordier, C. Aubert, M. Malacria, V. Gandon and E. Lacote, Chem.–Eur. J., 2010, 16, 9973 CrossRef CAS PubMed.
- M. F. Schlecht, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, New York, 1991, vol 7, p. 815 Search PubMed.
- J. Banerji, A. Chatterjee, N. Ghoshal, A. K. Das, S. Surkar, S. Bhattacharya and J. N. Shoolery, J. Indian Chem. Soc., 1982, 59, 145 CAS.
- J. Banerji, A. K. Das and B. Das, Chem. Ind., 1987, 395 CAS.
- H. Suginome and T. Uchida, J. Chem. Soc., Perkin Trans. 1, 1980, 943 RSC.
- H. Suginome, Y. Ohue and K. Orito, J. Chem. Soc., Perkin Trans. 1, 1987, 1247 RSC.
- Z. Wang, N. Shangguan, J. R. Cusick and L. Williams, J. Synlett., 2008, 213 Search PubMed.
- I. Fleming, J. H. M. Hill, D. Parker and D. Waterson, J. Chem. Soc., Chem. Commun., 1985, 318 RSC.
- Comprehensive Heterocyclic Chemistry, ed. G. V. Boyd, A. R. Katritzky and C. W. Rees, Pergamon, Oxford, 1984, vol. 6, ch. 4.18 Search PubMed.
- Comprehensive Heterocyclic Chemistry II, ed. F. W. J. r. Hartner, A. R. Katritzky, C. W. Rees and E. F. V. Scriven, Elsevier Science, Oxford, 1996, vol. 3, ch. 3.04 Search PubMed.
- G. Pattenden, J. Heterocycl. Chem., 1992, 29, 607 CrossRef CAS.
- J. P. Michael and G. Pattenden, Angew. Chem., Int. Ed. Engl., 1993, 32, 1 CrossRef.
- P. Wipf, Chem. Rev., 1995, 95, 2115 CrossRef CAS.
- E. Vedejs and S. D. Monahan, J. Org. Chem., 1996, 61, 5192 CrossRef CAS.
- Y. Kawano and H. Togo, Synlett, 2008, 217 CAS.
- T. Seko, S. Katsumata, M. Kato, J. I. Manako and K. Ohmoto, Application: WO 2003068753 A1 20030821, Ono Pharmaceutical Co., Ltd, Japan, 2003, p. 222.
- F. Waetjen, B. H. Dahl, J. Drejer and L. H. Jensen, Application: Cont-in-part of US 5 242 918, NeuroSearch A/S, Den., 1995, p. 8.
- H. Ogawa, K. Kondo, H. Yamashita, K. Kan, M. Tominaga and Y. Yabuuchi, Application: WO 9404525 A1 19940303, Otsuka Pharmaceutical Co., Ltd., Japan, 1994, p. 159 Search PubMed.
- L. Cui, G. Zhang, Y. Peng and L. Zhang, Org. Lett., 2009, 6, 1225 CrossRef PubMed.
- P. R. Hanson, D. A. Probst, R. E. Robinson and M. Yau, Tetrahedron Lett., 1999, 40, 4761 CrossRef CAS.
- W. R. Rough, S. L. Gwaltney, J. Cheng, K. A. Scheidt, J. H. McKerrow and E. Hansell, J. Am. Chem. Soc., 1998, 120, 10994 CrossRef.
- W. J. Moree, G. A. van der Marel and R. M. J. Liskamp, J. Org. Chem., 1995, 60, 5157 CrossRef CAS.
- C. Gennari, H. P. Nestler, B. Salom and W. C. Still, Angew. Chem., Int. Ed. Engl., 1995, 34, 1765 CrossRef CAS.
- C. Gennari, B. Salom, D. Potenza and A. Williams, Angew. Chem., Int. Ed. Engl., 1994, 33, 2067 CrossRef.
- W. J. Moree, L. C. van Gent, G. A. van der Marel and R. M. J. Liskamp, Tetrahedron, 1993, 49, 1133 CrossRef CAS.
- W. J. Moree, G. A. van der Marel and R. M. Liskamp, Tetrahedron Lett., 1991, 32, 409 CrossRef CAS.
- G. P. Zecchini, M. P. Paradisi, I. Torrini, G. Lucente, E. Gavuzzo, F. Mazza and G. Pochetti, Tetrahedron Lett., 1991, 32, 6779 CrossRef CAS.
- S. Hayashi, H. Ueki, Y. Sako, T. Ashunura, K. Hayashi and K. Takase, Kumamoto Pharm. Bull., 1962, 5, 51 CAS.
- H. Friebel and S. Sommer, Dtsch. Med. Wochenschr., 1960, 85, 2192 CrossRef CAS PubMed.
- K. H. Ahn, C. Ham, S. K. Kim and C. W. Cho, J. Org. Chem., 1997, 62, 7047 CrossRef CAS.
- W. Oppolzer, A. J. Kingma and S. K. Pillai, Tetrahedron Lett., 1991, 32, 4893 CrossRef CAS.
- W. Oppolzer, M. Wills, C. Starkemann and G. Bernardinelli, Tetrahedron Lett., 1990, 31, 4117 CrossRef CAS.
- W. Oppolzer, M. Wills, M. J. Kelly, M. Signer and J. Blagg, Tetrahedron Lett., 1990, 31, 5015 CrossRef CAS.
- W. Oppolzer, I. Rodriguez, C. Starkemann and E. Walther, Tetrahedron Lett., 1990, 31, 5019 CrossRef CAS.
- Y. Ishiwata and H. Togo, Tetrahedron Lett., 2009, 50, 5354 CrossRef CAS PubMed.
- J. Iqbal, A. Pandey and B. P. S. Chauhan, Tetrahedron, 1991, 41, 4143 CrossRef.
- C. H. Heathcock, S. L. Graham, M. C. Pirrung, F. Plavac, and C. T. White, in The Total Synthesis of Natural Products, ed. J. Apsimon, Wiley-Interscience, New York, 1983; vol. 5, p. 264 Search PubMed.
- H. Greger, Planta Med., 2006, 72, 99 CrossRef CAS PubMed.
- V. I. Minkin, Chem. Rev., 2004, 104, 2751 CrossRef CAS PubMed.
- R. Pudzich, T. Fuhrmann-Lieker and J. Salbeck, Adv. Polym. Sci., 2006, 199, 83 CrossRef CAS.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS PubMed.
- K. Ding, Z. Han and Z. Wang, Chem.–Asian J., 2009, 4, 32 CrossRef CAS PubMed.
- T. Dohi, T. Nakae, Y. Ishikado, D. Kato and Y. Kita, Org. Biomol. Chem., 2011, 9, 6899 CAS.
- K. Watanabe, Y. Iwata, S. Adachi, T. Nishikawa, Y. Yoshida, S. Kameda, M. Ide, Y. Saikawa and M. Nakata, J. Org. Chem., 2010, 75, 5573 CrossRef CAS PubMed.
- D. A. Henderson, P. N. Collier, G. Pave, P. Rzepa, A. J. P. White, J. N. Burrows and A. G. M. Barrett, J. Org. Chem., 2006, 71, 2434 CrossRef CAS PubMed.
- F. Konno, T. Ishikawa, M. Kawahata and K. Yamaguchi, J. Org. Chem., 2006, 71, 9818 CrossRef CAS PubMed.
- C. Cox and S. J. Danishefsky, Org. Lett., 2000, 2, 3493 CrossRef CAS PubMed.
- J. Hassan, M. Sevignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359 CrossRef CAS PubMed.
- E. Gellert, J. Nat. Prod., 1982, 45, 50 CrossRef CAS.
- Z. G. Li, Z. Jin and R. Q. Huang, Synthesis, 2001, 16, 2365 CrossRef PubMed.
- M. Suffness and G. A. Cordell, The Alkaloids. Chemistry and Pharmacology, ed. A. Brossi, Academic Press, Orlando, FL, 1985, vol. 25, p. 3 Search PubMed.
- Z. Jin, Z. G. Li and R. Q. Huang, Nat. Prod. Rep., 2002, 19, 454 RSC.
- K. Wang, M. Lu, A. Yu, X. Zhu and Q. Wang, J. Org. Chem., 2009, 74, 935 CrossRef CAS PubMed.
- R. M. Moriarty, R. K. Vaid and G. F. Koser, Synlett, 1990, 365 CrossRef CAS PubMed.
- G. F. Koser, Aldrichimica Acta, 2001, 34, 89 CAS.
- O. Prakash, N. Saini and P. K. Sharma, Heterocycles, 1994, 38, 409 CrossRef CAS PubMed.
- O. Neilands and B. Karele, J. Org. Chem., USSR, 1970, 6, 885 CAS.
- G. F. Koser, R. H. Wettach, J. M. Troup and B. A. Frenz, J. Org. Chem., 1976, 41, 3609 CrossRef CAS.
- G. F. Koser and R. H. Wettach, J. Org. Chem., 1977, 42, 1476 CrossRef CAS.
- G. F. Koser, R. H. Wettach and C. S. Smith, J. Org. Chem., 1980, 45, 1543 CrossRef CAS.
- G. F. Koser, A. G. Relenyi, A. N. Kalos, L. Rebrovic and R. H. Wettach, J. Org. Chem., 1982, 47, 2487 CrossRef CAS.
- R. M. Moriarty, R. Penmasta, A. K. Awasthi, W. R. Epa and I. Prakash, J. Org. Chem., 1989, 54, 1101 CrossRef CAS.
- R. M. Moriarty, R. K. Vaid, T. E. Hopkins, B. K. Vaid and O. Prakash, Tetrahedron Lett., 1990, 31, 201 CrossRef CAS.
- A. Tuncay, J. A. Dustman, G. Fisher and C. I. Tuncay, Tetrahedron Lett., 1992, 33, 7647 CrossRef CAS.
- R. M. Moriarty, B. K. Vaid, M. P. Duncan, S. G. Levy, O. Prakash and S. Goyal, Synthesis, 1992, 845 CrossRef CAS.
- O. Prakash and S. Goyal, Synthesis, 1992, 629 CrossRef CAS PubMed.
- O. Prakash, N. Rani and S. Goyal, J. Chem. Soc., Perkin Trans. 1, 1992, 707 RSC.
- O. Prakash, N. Saini and P. K. Sharma, Synlett, 1994, 221 CrossRef CAS PubMed.
- R. S. Vrama, D. Kumar and P. J. Liesen, J. Chem. Soc., Perkin Trans. 1, 1998, 4093 RSC.
- J. C. Lee and J. H. Choi, Synlett, 2001, 234 CAS.
- P. Lai and M. S. Taylor, Synthesis, 2010, 1449 CAS.
- Y. Yamamoto, Y. Kawano, P. H. Toy and H. Togo, Tetrahedron, 2007, 63, 4680 CrossRef CAS PubMed.
- Y. Yamamoto and H. Togo, Synlett, 2006, 798 CAS.
- Y. Suzuki and H. Togo, Synthesis, 2010, 2355 CAS.
- A. Tanaka, K. Moriyama and H. Togo, Synlett, 2011, 1853 CAS.
- J. Akiike, Y. Yamamoto and H. Togo, Synlett, 2007, 2168 CAS.
- R. D. Richardson, T. K. Page, S. Altermann, S. M. Paradine, A. N. French and T. Wirth, Synlett, 2007, 538 CAS.
- S. M. Altermann, R. D. Richardson, T. K. Page, R. K. Schmidt, E. Holland, U. Mohammed, S. M. Paradine, A. N. French, C. Richter, A. M. Bahar, B. Witulski and T. Wirth, Eur. J. Org. Chem., 2008, 5315 CrossRef CAS.
- H. Hamamoto, G. Anilkumar, H. Tohma and Y. Kita, Chem.–Eur. J., 2002, 8, 5377 CrossRef CAS.
- A. A. Guilbault, B. Basdevant, V. Wanie and C. Y. Legault, J. Org. Chem., 2012, 77, 11283 CrossRef CAS PubMed.
- M. Ochiai, Y. Takeuchi, T. Katayama, T. Sueda and K. Miyamoto, J. Am. Chem. Soc., 2005, 127, 12244 CrossRef CAS PubMed.
- C. W. Downey, S. Craciun, C. A. Vivelo, A. M. Neferu, C. J. Mueller and S. Corsi, Tetrahedron Lett., 2012, 53, 5766 CrossRef CAS PubMed.
- T. Thiemann, Y. Tanaka and J. Iniesta, Molecules, 2009, 14, 1013 CrossRef CAS PubMed.
- A. H. Katz, C. A. Demerson, C. C. Shaw, A. A. Asselin, L. G. Humber, K. M. Conway, G. Gavin, C. N. Guinosso, P. Jensen, D. Mobilio, R. Noureldin, J. Schmid, U. Shah, D. V. Engen, T. T. Chau and B. M. Weichman, J. Med. Chem., 1988, 31, 1244 CrossRef CAS.
- K. Higuchi, R. Takei, T. Kouko and T. Kawasaki, Synthesis, 2007, 669 Search PubMed.
- J. F. Liu, Z. Y. Jiang, R. R. Wang, Y. T. Zheng, J. J. Chen, X. M. Zhang and Y. B. Ma, Org. Lett., 2007, 9, 4127 CrossRef CAS PubMed.
- T. Kouko, K. Matsumura and T. Kawasaki, Tetrahedron, 2005, 61, 2309 CrossRef CAS PubMed.
- T. Kawasaki, H. Enoki, K. Matsumura, M. Ohyama, M. Inagawa and M. Sakamoto, Org. Lett., 2000, 2, 3027 CrossRef CAS PubMed.
- A. Casapullo, G. Bifulco, I. Bruno and R. Riccio, J. Nat. Prod., 2000, 63, 447 CrossRef CAS PubMed.
- R. J. Capon, F. Rooney, L. M. Murray, E. Collins, A. T. R. Sim, J. A. P. Rostas, M. S. Butler and A. R. Carroll, J. Nat. Prod., 1998, 61, 660 CrossRef CAS PubMed.
- Z. Xu, X. Yu, X. Feng and M. Bao, J. Org. Chem., 2012, 77, 7114 CrossRef CAS PubMed.
- A. M. Jones, G. Liu, M. M. Lorion, S. Patterson, T. Lebl, A. M. Z. Slawin and N. J. Westwood, Chem.–Eur. J., 2011, 17, 5714 CrossRef CAS PubMed.
- E. N. Prilezhaeva, Russ. Chem. Rev., 2000, 69, 367 CrossRef CAS PubMed.
- Z. Jin, P. C. Vandort and P. L. Fuchs, Phosphorus Sulfur Silicon, 1994, 95, 1 CrossRef.
- S. Patai and Z. Rappoport, Synthesis of Sulfones, Sulfoxides and Cyclic Sulfides, John Wiley, Chichester, 1994 Search PubMed.
- N. S. Simpkins, Sulfones in Organic Synthesis, Pergamon Press, Oxford, 1993 Search PubMed.
- G. Andrew, F. Ian, L. Andrew, P. Andrew, W. Graham and G. Vincenzo, European Patent EP., 1511727, 2007.
- G. A. Ellard, P. T. Gammon, H. S. Helmy and R. J. W. Rees, Nature, 1972, 239, 159 CrossRef CAS.
- T. F. Parsons, J. D. Buckman, D. E. Pearson and L. Field, J. Org. Chem., 1965, 30, 1923 CrossRef CAS.
- G. Palumbo, C. D. Ferreri, C. Ambrocio and R. Caputo, Phosphorus Sulfur Silicon, 1984, 19, 235 CAS.
- K. Fujiki and E. Y. Yoshida, Synth. Commun., 1999, 29, 3289 CrossRef CAS.
- K. Fujiki, S. Akieda, H. Yasuda and Y. Sasaki, Synthesis, 2001, 1035 CrossRef CAS PubMed.
- J. P. Weidner and S. S. Block, J. Med. Chem., 1964, 7, 671 CrossRef CAS.
- S. S. Block and J. P. Weidner, Mech. React. Sulfur Compd., 1967, 2, 235 Search PubMed.
- W. G. Filby, K. Guenther and R. D. Penzhorn, J. Org. Chem., 1973, 38, 4070 CrossRef CAS.
- B. M. Trost, T. N. Salzmann and K. Hiroi, J. Am. Chem. Soc., 1976, 98, 4887 CrossRef CAS.
- M. Madesclaire, Tetrahedron, 1986, 42, 5459 CrossRef CAS.
- L. Dai, D. Fan, X. Wang and Y. Chen, Synth. Commun., 2008, 38, 576 CrossRef CAS.
- V. G. Nenajdenko, A. L. Krasovskiy and E. S. Balenkova, Tetrahedron, 2007, 63, 12481 CrossRef CAS PubMed.
- T. Nagata, T. Yoshino, N. Haginoya, K. Yoshikawa, M. Nagamochi, S. Kobayashi, S. Komoriya, A. Yokomizo, R. Muto, M. Yamaguchi, K. Osanai, M. Suzuki and H. Kanno, Bioorg. Med. Chem., 2009, 17, 1193 CrossRef CAS PubMed.
- L. Varoli, P. Angeli, S. Burnelli, G. Marucci and M. Recanatini, Bioorg. Med. Chem., 1999, 7, 1837 CrossRef CAS.
- S. Canova, R. Lépine, A. Thys, A. Baron and D. Roche, Bioorg. Med. Chem. Lett., 2011, 21, 4768 CrossRef CAS PubMed.
- G. L. Khatik, J. Kaur, V. Kumar, K. Tikoo and V. A. Nair, Bioorg. Med. Chem. Lett., 2012, 22, 1912 CrossRef CAS PubMed.
- M. Sakairi, M. Kogami, M. Torii, H. Kataoka, H. Fujieda, M. Makino, D. Kataoka, R. Okamoto, T. Miyazawa, M. Okabe, M. Inoue, N. Takahashi, S. Harada and N. Watanabe, Bioorg. Med. Chem. Lett., 2012, 22, 5123 CrossRef CAS PubMed.
- H. S. Rho, D. S. Yoo, S. M. Ahn, M. Y. Kim, D. H. Cho and J. Y. Cho, Bull. Korean Chem. Soc., 2010, 31, 3463 CrossRef CAS.
- A. Thompson, J. R. Garabatos-Perera and H. M. Gillis, Can. J. Chem., 2008, 86, 676 CrossRef CAS.
- E. Wojaczynska and J. Wojaczynski, Chem. Rev., 2010, 110, 4303 CrossRef CAS PubMed.
- T. Shigetomi, H. Soejima, Y. Nibu, K. Shioji, K. Okuma and Y. Yokomori, Chem.–Eur. J., 2006, 12, 7742 CrossRef CAS PubMed.
- I. Potorocina, M. Vorona, I. Shestakova, I. Domracheva, E. Liepinsh and G. Veinberg, Chem. Heterocycl. Compd., 2011, 47, 767 CrossRef CAS.
- A. V. Timshina, S. A. Rubtsova, N. Alekseev, M. I. Kodess, E. G. Mamochkina, P. A. Slepukhin and A. V. Kuchin, Chem. Nat. Compd., 2008, 44, 728 CrossRef CAS.
- J. Wu, W. Zhao and S. Cao, Eur. J. Org. Chem., 2012, 1380 CrossRef CAS.
- J. R. Fotsing and K. Banert, Eur. J. Org. Chem., 2005, 3704 CrossRef CAS.
- I. M. Rafiqul, K. Shimada, S. Aoyagi, Y. Takikawa and C. Kabuto, Heteroat. Chem., 2004, 15, 165 Search PubMed.
- R. J. Griffin, A. Henderson, N. J. Curtin, A. Echalier, J. A. Endicott, I. R. Hardcastle, D. R. Newell, M. E. M. Noble, L. Z. Wang and B. T. Golding, J. Am. Chem. Soc., 2006, 128, 6012 CrossRef CAS PubMed.
- M. A. Brimble and E. Ireland, J. Chem. Soc., Perkin Trans. 1, 1994, 3109 RSC.
- I. Iwama, H. Matsumoto, H. Shimizu, T. Kataoka, O. Muraoka and G. Tanabe, J. Chem. Soc., Perkin Trans. 1, 1998, 1569 RSC.
- M. J. Lai, H. L. Huang, S. L. Pan, Y. M. Liu, C. Y. Peng, H. Y. Lee, T. K. Yeh, P. H. Huang, C. M. Teng, C. S. Chen, H. Y. Chuang and J. P. Liou, J. Med. Chem., 2012, 55, 3777 CrossRef CAS PubMed.
- A. Treiber, J. Org. Chem., 2002, 67, 7261 CrossRef CAS PubMed.
- F. Freeman and C. N. Angeletakis, J. Am. Chem. Soc., 1983, 105, 4039 CrossRef CAS.
- A. K. Bhattacharya and A. G. Hortmann, J. Org. Chem., 1978, 43, 2728 CrossRef CAS.
- J. M. Salvino, S. Patel, M. Drew, P. Krowlikowski, E. Orton, N. V. Kumar, T. Caulfield and R. Labaudiniere, J. Comb. Chem., 2001, 3, 177 CrossRef CAS PubMed.
- K. Ramasamy, N. Imamura, N. B. Hanna, R. A. Finch, T. L. Avery, R. K. Robins and G. R. Revankar, J. Med. Chem., 1990, 33, 1220 CrossRef CAS.
- Y. Asahina, K. Iwase, F. Iinuma, M. Hosaka and T. Ishizaki, J. Med. Chem., 2005, 48, 3194 CrossRef CAS PubMed.
- F. Freeman and C. N. Angeletakis, J. Org. Chem., 1985, 50, 793 CrossRef CAS.
- J. L. Kice, L. Weclas-Henderson and A. Kewan, J. Org. Chem., 1989, 54, 4198 CrossRef CAS.
- X. Zhu, Y. Jin and J. Wickham, J. Org. Chem., 2007, 72, 2670 CrossRef CAS PubMed.
- D. B. Werz, C. Bleiholder, R. Gleiter and F. Rominger, J. Organomet. Chem., 2006, 691, 3943 CrossRef CAS PubMed.
- W. N. Wu, L. A. McKown, K. A. Yorgey and J. F. Pritchard, J. Pharm. Biomed. Anal., 1999, 20, 687 CrossRef CAS.
- H. Kuroda, I. Tomita and T. Endo, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 2589 CrossRef CAS.
- K. L. Stensaas, A. S. Brownell, S. Ahuja, J. K. Harriss and S. R. Herman, J. Sulfur Chem., 2008, 29, 433 CrossRef CAS.
- R. Priefer, E. Martineau and D. N. Harpp, J. Sulfur Chem., 2007, 28, 529 CrossRef CAS.
- C. D. Gabbutt, B. M. Heron, S. B. Kolla, C. Kilner, S. J. Coles, P. N. Horton and M. B. Hursthouse, Org. Biomol. Chem., 2008, 6, 3096 CAS.
- Y. Suzuki, T. Okamoto, A. Wakamiya and S. Yamaguchi, Org. Lett., 2008, 10, 3393 CrossRef CAS PubMed.
- O. G. Mancheno, O. Bistri and C. Bolm, Org. Lett., 2007, 9, 3809 CrossRef PubMed.
- A. M. Bernard, A. Frongia, P. P. Piras, F. Secci and M. Spiga, Org. Lett., 2005, 7, 4565 CrossRef CAS PubMed.
- A. P. Avdeenko, S. A. Konovalova and A. A. Santalova, Russ. J. Org. Chem., 2008, 44, 231 CrossRef CAS.
- A. N. Galyautdinova, R. M. Vafina, O. N. Kataeva, O. A. Lodochnikova, S. G. Gnevashev, O. I. Gnezdilov, V. V. Gavrilov, Y. G. Shtyrlin, G. A. Chmutova and E. N. Klimovitskii, Russ. J. Org. Chem., 2010, 46, 246 CrossRef CAS.
- R. D. Larsen and F. E. Roberts, Synth. Commun., 1986, 16, 899 CrossRef CAS.
- F. Fringuelli, R. Pellegrino, O. Piermatti and F. Pizzo, Synth. Commun., 1994, 24, 2665 CrossRef CAS.
- M. D. Rosen, M. D. Doubleday and M. J. Suto, Synth. Commun., 1998, 28, 3491 CrossRef CAS.
- K. H. Yoo, S. E. Kim, K. J. Shin, D. C. Kim, S. W. Park and D. J. Kim, Synth. Commun., 2001, 31, 835 CrossRef CAS PubMed.
- D. S. Perlow, M. S. Kuo, H. M. Moritz, J. S. Wai and M. S. Egbertson, Synth. Commun., 2001, 37, 1887 CrossRef.
- J. L. G. Ruano, A. Parra, F. Yuste and V. M. Mastranzo, Synthesis, 2008, 311 CrossRef CAS PubMed.
- A. M. Le Nocher and P. Metzner, Tetrahedron Lett., 1991, 32, 747 CrossRef CAS.
- U. Ellervik, M. Jacobsson and J. Ohlsson, Tetrahedron, 2005, 61, 2421 CrossRef CAS PubMed.
- G. Mloston, K. Urbaniak, A. Linden and H. Heimgartner, Tetrahedron, 2009, 65, 8191 CrossRef CAS PubMed.
- P. K. Dornan, P. L. Leung and V. M. Dong, Tetrahedron, 2011, 67, 4378 CrossRef CAS PubMed.
- A. Mukherjee and N. Jayaraman, Tetrahedron, 2012, 68, 8746 CrossRef CAS PubMed.
- H. Yuasa and H. Hashimoto, Tetrahedron, 1993, 49, 8977 CrossRef CAS.
- A. C. Tome, J. A. S. Cavaleiro and R. C. Storr, Tetrahedron, 1996, 52, 1723 CrossRef CAS.
- A. C. Tome, J. A. S. Cavaleiro and R. C. Storr, Tetrahedron, 1996, 52, 1735 CrossRef CAS.
- C. Leriverend, P. Metzner, A. Capperucci and A. D. Innocenti, Tetrahedron, 1997, 53, 1323 CrossRef CAS.
- H. N. Tran and T. N. Pham, Tetrahedron, 2001, 57, 1289 CrossRef CAS.
- A. K. Mohanakrishnan and N. Ramesh, Tetrahedron Lett., 2005, 46, 4231 CrossRef CAS PubMed.
- J. Nakayama, A. Mizumura, Y. Yokomori, A. Krebs and K. Schutz, Tetrahedron Lett., 1995, 36, 8583 CrossRef CAS.
- G. Capozzi, A. Corti, S. Menichetti and C. Nativi, Tetrahedron Lett., 1997, 38, 5041 CrossRef CAS.
- M. S. Kim and I. H. Jeong, Tetrahedron Lett., 2005, 46, 3545 CrossRef CAS PubMed.
- M. Sekine, S. Iimura and T. Nakanishi, Tetrahedron Lett., 1991, 32, 395 CrossRef CAS.
- P. M. Cullis, Chem. Commun., 1984, 1510 RSC.
- C. P. Chow and C. E. Berkman, Tetrahedron Lett., 1998, 39, 7471 CrossRef CAS.
- A. Toshimitsu, C. Hirosawa, S. Tanimoto and S. Uemura, Tetrahedron Lett., 1992, 58, 322 Search PubMed.
- R. L. Danheiser, Y. M. Choi, M. Menichincheri and E. J. Stoner, J. Org. Chem., 1993, 58, 322 CrossRef CAS.
- H. J. Reich and S. K. J. Shah, J. Am. Chem. Soc., 1975, 97, 3250 CrossRef CAS.
- H. Hussain, I. R. Green and I. Ahmed, Chem. Rev., 2013, 113, 3329–3371 CrossRef CAS PubMed.
- M. H. Lim, Y. J. Lee, Y. M. Goh, W. Nam and C. Kim, Bull. Chem. Soc. Jpn., 1999, 72, 707 CrossRef CAS.
- T. Konoike, Y. Araki and Y. Kanda, Tetrahedron Lett., 1999, 40, 6971 CrossRef CAS.
- A. Varvoglis, Hypervalent Iodine in Organic Synthesis, Academic Press, San Diego, 1997 Search PubMed.
- Y. Yamamoto and H. Togo, Synlett, 2005, 2486 CAS.
- E. A. Merritt, V. M. T. Carneiro, L. F. Silva Jr and B. Olofsson, J. Org. Chem., 2010, 75, 7416 CrossRef CAS PubMed.
- M. Uyanik, T. Yasui and K. Ishihara, Bioorg. Med. Chem. Lett., 2009, 19, 3848 CrossRef CAS PubMed.
- M. Nandakumar, R. Sivasakthikumaran and A. K. Mohanakrishnan, Eur. J. Org. Chem., 2012, 3647 CrossRef CAS.
- B. Witkop and H. Fiedler, Justus Liebigs Ann. Chem., 1947, 558, 91 CrossRef.
- T. Hino, H. Yamaguchi, K. Matsuki, K. Nakano, S. Sodeoka and M. J. Nakagawa, J. Chem. Soc., Perkin Trans. 1, 1983, 141 RSC.
- R. Güller and H. J. Borschberg, Helv. Chim. Acta, 1993, 76, 1847 CrossRef.
- R. J. Friary and J. H. Schwerdt, Tetrahedron, 1991, 47, 9981 CrossRef CAS.
- T. Kurihara, Y. Sakamoto, M. Takai, H. Ohishi, S. Harusawa and R. Yoneda, Chem. Pharm. Bull., 1995, 43, 1089 CrossRef CAS.
- C. Tratrat, S. Giorgi-Renault and H. P. Husson, J. Org. Chem., 2000, 65, 6773 CrossRef CAS.
- M. Góra, M. K. Luczynski and J. J. Sepiol, Synthesis, 2005, 1625 CrossRef PubMed.
- J. Y. Kim, H. Rhee, M. Kim and J. Korean, Chem. Soc., 2002, 46, 479 CAS.
- K. S. Kochharl, D. A. Cottrell and H. W. Pinnick, Tetrahedron Lett., 1983, 24, 1323 CrossRef.
- T. Inokuchi and H. Kawafuchi, Tetrahedron, 2004, 60, 11969 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.