
Effect of sodium acetate in atom transfer radical addition of polyhaloalkanes to olefins†
Abstract
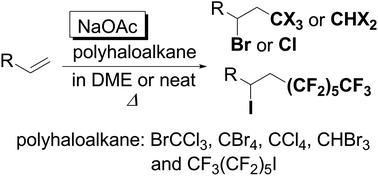
The atom transfer radical addition of polyhaloalkanes, such as bromotrichloromethane and polyfluoroalkyl iodine, to olefins smoothly proceeds in the presence of sodium acetate as an efficient auxiliary agent in dimethoxyethane. The present transition metal- and peroxide-free methodology is applicable to a broad scope of substrates.
 Please wait while we load your content...
Please wait while we load your content...

