
Mesoporous silica: a highly promising and compatible candidate for optical and biomedical applications†
Abstract
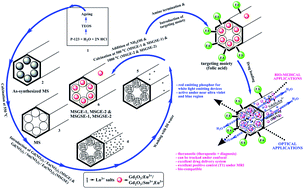
A simple post-co-precipitation strategy is adopted for the construction of a mesoporous silica framework in which the rare earth gadolinium oxide (Gd2O3:Eu3+& Gd2O3:Sm3+, Eu3+) is incorporated as an emissive probe for optical and bio-medical applications. The samples show good emission along with the required theranostic properties. In the blue region, a better red emissive behavior is observed in the case of Eu3+ doped sample while the Sm3+ and Eu3+ co-doped sample exhibit the same in the near ultra-violet region. Moreover, the structural and textural properties of mesoporous silica are preserved even in the presence of rare earth doped Gd2O3 and this can very well be utilized for theranostic applications. A clear confocal microscopy image shows a red spot, and thus its potential towards trackability is confirmed. In addition to this, other admirable properties are also associated with it namely, an excellent drug loading ability, bio-compatibility, easy surface modification by the targeting moiety and excellent longitudinal relaxivity (T1) under magnetic resonance imaging (MRI), which stands par for it to be utilized in the field of theranostics too. This material shows good luminescence properties along with the desired internal–external morphology, and a narrowly ordered porosity can validate the application of these materials in multi-faceted fields such as phosphors for LEDs, bio-imaging, theranostics etc.
 Please wait while we load your content...
Please wait while we load your content...

