Reduction with tris(2-carboxyethyl)phosphine (TCEP) enables the use of an S-sulphonate protecting group for thiol-mediated bioconjugation†
Barbara Maretab,
Thomas Regnierb,
Jean-Christophe Rossia,
Laurent Garrellyb,
Laurent Viala and
Robert Pascal*a
aInstitut des Biomolécules Max Mousseron, UMR 5247 CNRS – Universités Montpellier 1 & Montpellier 2, CC17006, Place E. Bataillon, F-34095, Montpellier, France. E-mail: rpascal@univ-montp2.fr
bCOLCOM SARL, Cap Alpha Avenue de l'Europe, F-34830, Clapiers, France
First published on 10th January 2014
Abstract
Herein, we demonstrate the effectiveness of the water-friendly S-sulphonate group as an alternative to traditional thiol protecting groups for subsequent deprotection–bioconjugation reactions, under conditions that are compatible with the use of biochemical samples.
In the course of our on-going studies on the functionalization of poly-L-lysine dendrigrafts,1 we were interested in the synthesis of thiol-containing entities for bioconjugation reactions in aqueous media. Due to the hydrophobicity of the traditional thiol-protecting groups such as S-trityl,2 which usually leads to low water-soluble species, we envisioned the alternative use of the water-friendly anionic S-sulphonate function. The thiosulphate moiety is a masked thiol and is stable enough to endure further synthetic steps. For example, S-sulphocysteine derivatives have been used during peptide synthesis,3 the preparation of carbapenem antibiotics,4 RAFT polymerisation reactions,5 the preparation of self-assembled monolayers on gold surfaces,6 or reactions with isobenzofuranone and isoindolone carbocations.7
S-Sulphonates, also known as Bunte salts or S-alkyl thiosulphates, can be efficiently prepared either by oxidation of thiols in presence of sulphites,8 or by nucleophilic substitution of alkyl halides with thiosulphate (Scheme 1).9 However, deprotection reactions from the Bunte salts in aqueous solutions reported so far occur under strong acidic conditions,10 by exchange reactions in the presence of high concentrations of other thiols,9 or by reduction with NaBH4.11 These conditions are likely to limit their use for delicate substrates such as proteins or nucleic acids.
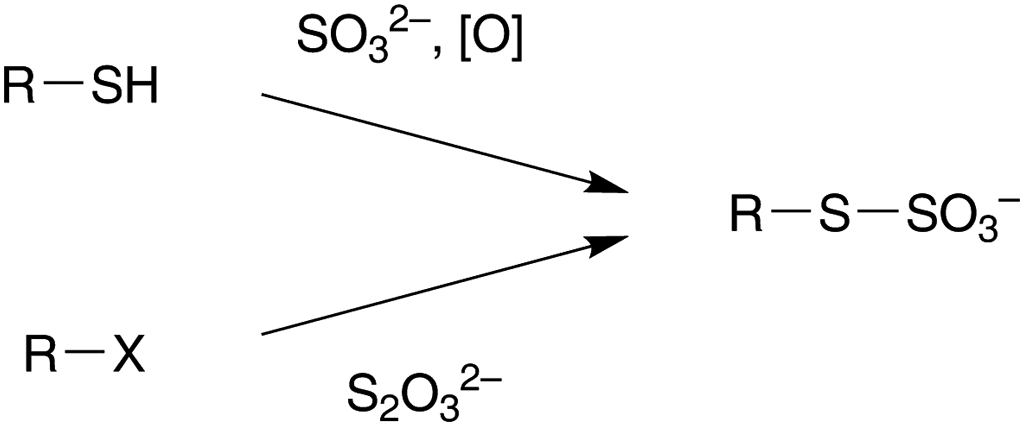 | ||
| Scheme 1 Formation of S-sulphonates derivatives (Bunte salts). | ||
In this communication, we report the unprecedented use of TCEP (tris(2-carboxyethyl)phosphine)12 and agarose-immobilized TCEP, an odourless and selective reducing reagent,13 for the removal of the S-sulphonate protection releasing free thiols in neutral aqueous media. The compatibility of the deprotection conditions with a subsequent nucleophilic addition of the free thiols on a model maleimide substrate is demonstrated. Finally, this deprotection–ligation procedure was applied to the formation of a bioconjugate between fluorescein and horseradish peroxidase (HRP).
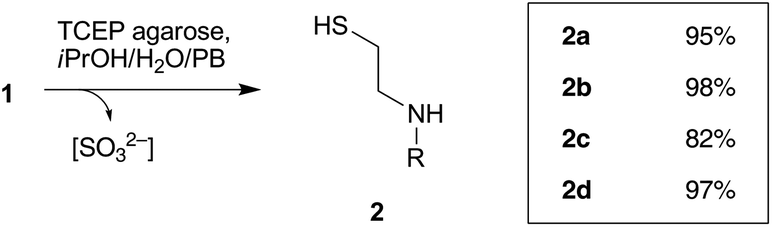
Substrates 1a–d that bear different molecular tags – in order to monitor the reaction progress or that are relevant in a bioconjugation framework – were easily prepared by acylation or sulphonylation reactions from the commercially available S-(2-aminoethyl)thiosulphuric acid (49–74% yield, Scheme 2).14 Bunte salts 1a–d were then mixed with immobilized TCEP on agarose beads in a iPrOH–phosphate buffer 0.5 M mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9, pH 7.4) at room temperature. The agarose beads were removed by centrifugation and the mother liquors were analyzed by HPLC-MS (Fig. 1 and ESI†), showing good to excellent conversions towards thiols 2a–d (82–97%, Scheme 3). The use of free TCEP led to similar conversion yields as indicated by the HPLC monitoring of the reaction medium (ESI).†
:9, pH 7.4) at room temperature. The agarose beads were removed by centrifugation and the mother liquors were analyzed by HPLC-MS (Fig. 1 and ESI†), showing good to excellent conversions towards thiols 2a–d (82–97%, Scheme 3). The use of free TCEP led to similar conversion yields as indicated by the HPLC monitoring of the reaction medium (ESI).†
 | ||
| Scheme 2 Starting materials 1a–1d prepared for the study of TCEP-mediated reduction. Reaction conditions: 1a S-(2-aminoethyl)thiosulphuric acid (1 eq.), dansyl chloride (1.3 eq.), Et3N (2 eq.), MeCN, r.t., 18 h, 74%. 1b S-(2-aminoethyl)thiosulphuric acid (1 eq.), BzOSu (1 eq.), DMF, r.t., 18 h, 72%. 1c (a) S-(2-aminoethyl)thiosulphuric acid (1 eq.), 5(6)-carboxyfluorescein diacetate N-succinimidyl ester (1 eq.), Et3N (2.2 eq), DMF, r.t., 18 h, 50%; (b) removal of acetyl protecting groups: aq. (NH4)2CO3, r.t., 18 h, 100%. 1d S-(2-aminoethyl)thiosulphuric acid (1 eq.), biotin N-succinimidyl ester (1 eq.), Et3N (2 eq.), DMF, r.t., 18 h, 72%. | ||
 | ||
| Fig. 1 HPLC monitoring (detection at 286 nm) of the reaction of 1a with resin-bound TCEP: (a) before the addition of the reducing agent; (b) after 30 min reaction. Reaction conditions: 1a (1 μmol in iPrOH, 1 eq) was introduced in suspension of agarose-beaded TCEP (4 μmol) in 0.5 M phosphate buffer pH 7.4 (1 mL). Analysis conditions: see Scheme 3. | ||
 | ||
| Scheme 3 Removal of S-sulphonate protecting group from models 1a–1d with TCEP. Reaction conditions: 1a–1d (1 μmol), agarose beaded TCEP (4 μmol), iPrOH/0.5 M phosphate buffer pH 7.4 1/9, r.t., 30 min. Conversion yield determined by the area of HPLC peaks compared to the HPLC analysis of the starting material. Analysis conditions: XTerra MS Column (C18, 5 μm, 2.1 × 150 mm), eluent: gradient from 100% H2O/0.1% TFA to 100% MeCN/0.1% TFA over 20 min, 0.2 mL min−1. | ||
In order to assess the compatibility of the above deprotection conditions with a subsequent ‘thiol–ene’ Michael addition reaction,15 a two-fold excess of the (R)-N-(1-phenylethyl)maleimide 3 in iPrOH was added to the above crude reaction mixtures (Scheme 4). Thiols 2a–d were fully converted in one hour to the corresponding Michael addition products 4a–d (as shown by the absence of peak corresponding to 2a – retention time 15.8 min – in Fig. 2, see ESI† for the full results with respect to compounds 2b–d). By contrast, no ligation reaction took place between Bunte salts 1a–d and 3. The formation of the adducts 4a–d was concomitant with the formation of a side product (Fig. 2a, HPLC retention time: 12.1 min), which was identified by negative-ion mode ESI-MS as the sulphite–maleimide adduct 5 (Scheme 5). This observation was consistent with the well-known Michael addition of sulphite on olefins bearing electron withdrawing substituents,16,17 and is indicative that both the free thiol and sulphite are produced during the TCEP-mediated reduction of S-sulphonates. Hence, the deprotection reaction follows the reverse pathway of the oxidative protection of thiols as S-sulphonates (Scheme 1).18 The prior elimination of sulphite can be performed by solid-phase extraction (SPE), the latter procedure being straightforward and routinely used for protein purification (Fig. 2b).
 | ||
| Scheme 4 Michael addition of thiols 2a–d. Reaction conditions: 2a–2d (1 μmol), 3 (2 μmol), iPrOH/0.5 M phosphate buffer pH 7.4 (1/9, 1 mL), r.t., 1 h. Analysis conditions: see Scheme 3. | ||
 | ||
| Fig. 2 HPLC monitoring (detection at 214 nm) of the Michael addition of thiol 2a on maleimide 3: (a) without prior purification of 2a; (b) with prior extraction of 2a on a SPE cartridge. Reaction conditions: 2a (1 μmol), 3 (2 μmol), iPrOH/0.5 M phosphate buffer pH 7.4, 1:9 (1 mL). Analysis conditions: see Scheme 3. | ||
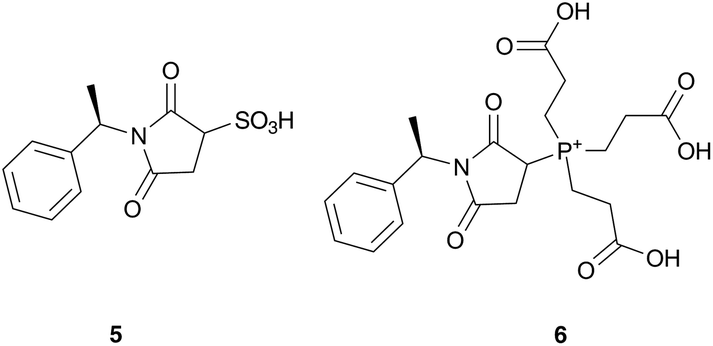 | ||
| Scheme 5 Side-products formed by Michael addition of sulphite and TCEP on maleimide 3. | ||
A second side product – identified as the maleimide adduct 6 – was formed when using free TCEP instead of its polymer-bound version (see ESI†), highlighting the advantage of using its polymer-bound version. The formation of 6 may result from the nucleophilic addition of excess TCEP on the Michael acceptor 3.
Finally, to confirm that biomolecules are not inactivated by the deprotection–ligation process we checked the effect of the procedure on biological activity. To this aim, the fluorescent S-sulphonate derivative 1c and maleimide-activated horseradish peroxidase (mal-HRP) were submitted to the whole chemical process. A final purification step using size exclusion chromatography (Fig. 3a) provided the fluorescein–horseradish peroxidase bioconjugate (fluo–HRP) as confirmed by its intense fluorescence properties vs. mal-HRP (RFUfluo–HRP/RFUmal-HRP = 153 ± 6, 80 μg mL−1, 5 mM phosphate buffer pH 7.4 and 150 mM NaCl, λexc 485 nm, λem 535 nm). We also verified that the enzyme integrity was neither altered by the presence of the covalently attached fluorescein, nor by the deprotection and conjugation reaction conditions (Fig. 3b) through monitoring the HRP-catalysed oxidation of o-phenylenediamine by UV spectroscopy according to a previously reported method.19 In spite of the fact that the above mentioned identification of side-products shows that the separation of sulphite and excess TCEP (or preferably the use of its resin-bound alternative) is preferable, this experiment also indicates that the chemical process is not harmful to biological activity and that a “one-pot” procedure can be acceptable in bioconjugation when pure adducts are not needed. In other instances, separation can be easily performed through SPE or liquid chromatography.
 | ||
| Fig. 3 (A) Size exclusion chromatograms for the Michael addition of 2c to mal-HRP. Column: Superdex 200 10/300GL; eluent: 5 mM phosphate buffer and 150 mM NaCl pH 7.4, flow 0.5 mL min−1; UV detection at 403 nm; fractions between 14 and 17 mL were collected. (B) Normalized HRP enzymatic activities. HRP concentrations in each collected fraction was determined using a micro BCA protein assay kit (Pierce, ref. 23235). | ||
In summary, we have demonstrated that the S-sulphonate function can be efficiently reduced with TCEP to afford unprotected sulfhydryl derivatives under reaction conditions that are compatible with a subsequent ‘thiol–ene’ Michael addition reaction and with the use of biomolecules. This procedure is likely to meet with success in the formation of bioconjugates through other thiol-mediated coupling reactions. Further investigations in this direction are currently underway.
Acknowledgements
The authors gratefully acknowledge the Agence Nationale de la Recherche et de la Technologie for financial support to Barbara Maret.Notes and references
- J.-C. Rossi, L. Boiteau, H. Collet, B. M. Tsamba, N. Larcher and R. Pascal, Tetrahedron Lett., 2012, 53, 2976–2979 CrossRef CAS PubMed; H. Collet, E. Souaid, H. Cottet, A. Deratani, L. Boiteau, G. Dessalces, J.-C. Rossi, A. Commeyras and R. Pascal, Chem. – Eur. J., 2010, 16, 2309–2316 CrossRef PubMed; A. Molero Bondia, N. Larcher, L. Garrelly, J. C. Rossi and R. Pascal, Tetrahedron Lett., 2010, 51, 3330–3333 CrossRef PubMed.
- Along with the p-methoxybenzyl thioether and S-2-trimethylsilylethyl protecting groups, see: P. G. M. Wuts and T. Greene, Greene's Protective Groups in Organic Synthesis, Wiley, Hoboken, NJ, 4th edn, 2007, ch. 6, p. 654 & p. 680 Search PubMed.
- Y. Sohma and S. B. H. Kent, J. Am. Chem. Soc., 2009, 131, 16313–16318 CrossRef CAS PubMed; S. Takeshi and S. Aimoto, Tetrahedron Lett., 2003, 44, 8085–8087 CrossRef PubMed; I. Maugras, J. Gosteli, E. Rappand and R. Nyfeler, Int. J. Pept. Protein Res., 1995, 45, 152–156 CrossRef.
- T. Isoda, I. Yamamura, S. Tamai, T. Kumaga and Y. Nagao, Chem. Pharm. Bull., 2006, 54, 1408–1411 CrossRef CAS.
- J. Jiang, X. Jia, X. Chen, Y. Zong, H. Zhang, L. Lin, X. Liu and M. Chen, Chem. Lett., 2011, 1378–1380 CrossRef CAS.
- J. Lukkari, M. Meretoja, I. Kartio, K. Laajalehto, M. Rajamäki, M. Lindström and J. Kankare, Langmuir, 1999, 15, 3529–3537 CrossRef CAS.
- L. Y. Ukhin, A. R. Akopova, A. V. Bicherov, L. G. Kuzmina, A. S. Morkovnik and G. S. Borodkin, Tetrahedron Lett., 2011, 52, 5444–5447 CrossRef CAS PubMed.
- P. G. M. Wuts and T. Greene, Greene's Protective Groups in Organic Synthesis, Wiley, Hoboken, NJ, 4th edn, 2007, ch. 6, p. 689 Search PubMed.
- J. March, Advanced Organic Chemistry, Wiley, New York, 4th edn, 1992, ch. 10, p. 410 CrossRef CAS; H. Distler, Angew. Chem., Int. Ed., 1967, 6, 544–553 CrossRef CAS.
- J. L. Kice, J. M. Anderson and N. E. Pawlowski, J. Am. Chem. Soc., 1966, 88, 5245–5250 CrossRef CAS; J. L. Kice, J. Org. Chem., 1963, 28, 957–961 CrossRef.
- J. C. P. Schwartz and K. C. Yule, Proc. Chem. Soc., London, 1961, 417 CAS.
- M. W. Jonesa, G. Mantovanib, S. M. Ryanc, X. Wangc, D. J. Braydenc and D. M. Haddleton, Chem. Commun., 2009, 5272–5274 RSC; A.-M. Faucher and C. Grand-Maître, Synth. Commun., 2003, 33, 3503–3511 CrossRef CAS PubMed; J. A. Burns, J. C. Butler, J. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef.
- G. Miralles, P. Verdié, K. Puget, A. Maurras, J. Martinez and G. Subra, ACS Comb. Sci., 2013, 15, 169–173 CrossRef CAS PubMed.
- See ESI for experimental details.†.
- A. Marra and A. Dondoni, Chem. Soc. Rev., 2012, 41, 576–586 Search PubMed; A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC; C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- A. Sakumoto, T. Miyata and M. Washino, Bull. Chem. Soc. Jpn., 1976, 49, 3584–3588 CrossRef CAS; H. D. Finch, J. Org. Chem., 1962, 27, 649–651 CrossRef.
- For instance, acridine derivatives of maleimide are used for the quantification of sulphite in biological samples, see: H. Meguro and H. Ohrui, Biosci., Biotechnol., Biochem., 1996, 60, 1919–1924 CrossRef CAS; K. Akasaka, H. Matsuda, H. Ohrui, H. Meguro and T. Suzuki, Agric. Biol. Chem., 1990, 54, 501–504 CrossRef.
- D. E. Shafer, J. K. Inman and A. Lees, Anal. Biochem., 2000, 282, 161–164 CrossRef CAS PubMed.
- T. Moreau, C. Faye, M. Baqué, I. Desvignes, G. Coussot, R. Pascal and O. Vandenabeele-Trambouze, Anal. Chim. Acta, 2011, 706, 354–360 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra47407k |
| This journal is © The Royal Society of Chemistry 2014 |