 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThree-step assembly of 4-aminotetrahydropyran-2-ones from isoxazoline-2-oxides†
A. S. Naumovaab,
A. A. Mikhaylov*a,
Yu. A. Khomutovaa,
R. A. Novikova,
D. E. Arkhipovc,
A. A. Korlyukovc and
S. L. Ioffe*a
aN. D. Zelinsky Institute of Organic Chemistry, Leninsky prosp. 47, 119991 Moscow, Russian Federation. E-mail: iof@ioc.ac.ru; Fax: +7-499-1355328
bHigher Chemical College of the Russian Academy of Sciences, Miusskaya sq. 9, 125047, Moscow, Russian Federation
cA. N. Nesmeyanov Institute of Organoelement Compounds, Vavilova str. 28, 119991 Moscow, Russian Federation
First published on 8th January 2014
Abstract
Tetrahydropyran-2-ones with a 4-amino function connected to a tertiary carbon atom – a widely naturally occurring fragment – are constructed by a three step protocol from easily available isoxazoline 2-oxides. In the first stage, the carbon skeleton of the target product is formed upon a C,C-coupling of a silyl ketene acetal with a nitronate function, under silyl triflate catalysis. The key step of the assembly consists of the oxidative cleavage of an endocyclic N–O bond of intermediate cyclic nitroso acetals with mCPBA, accompanied with lactone ring closure, and gives rise to β-nitro-δ-lactones in 63–85% yields. The latter are reduced with amalgamated aluminium, to furnish the target scaffold.
Introduction
The 4-aminosubstituted six-membered lactone scaffold (marked as structure 1 in Fig. 1), derived from β-amino acids, occurs in different classes of natural products, like stephadiamine,1 kopsihainin D2 and the tetrodotoxin (TTX) family3,4 (Fig. 1), exhibiting various biological activities. In particular, TTX and its congeners constitute the key components of the puffer fish fugu poison5 and due to their complex structure are considered as classical objects for total synthesis.6 However, few syntheses of these compounds have succeeded.7–10 Difficulties usually arise from the construction of the stereogenic quaternary carbon center connected with an amine (further guanidine) function. For this purpose, non-trivial or indirect methods such as Beckmann7b and Overman8 rearrangements, asymmetric transferring Strecker synthesis9 or chiral Rh-catalyzed C–H nitrene insertion10 have to be employed. Therefore, the development of simple, direct methods for the construction of lactones 1 is an urgent task. | ||
| Fig. 1 Selected natural products containing β-alkyl-β-amino-δ-valerolactone moiety 1. | ||
In this manuscript a convenient strategy for the synthesis of the scaffold 1 is suggested, which is based on a detailed investigation of the transformations of cyclic nitronates.11
A common retrosynthetic analysis for the β-amino acid moiety in lactones 1 employs a Mannich-type disconnection with the γ-imino alcohol cations A as key precursors for the generation of amines 1 (Scheme 1). However, this type of reaction is not generally applied, due to the complexity of the Mannich reaction with ketimines,12 although the approach could provide the fast and simple assembly of the target molecule 1.
 | ||
| Scheme 1 Retrosynthesis via Mannich-disconnection of scaffold 1. | ||
Our recent results in the area of five-membered cyclic nitronates chemistry13 suggest that the easily available five-membered nitronates 2 (isoxazoline 2-oxides)11 can serve as useful synthetic equivalents for ketiminium cations A (Scheme 1) in the reaction with a silyl ketene acetal, but this approach requires an additional step for the reduction of both of the N–O bonds and the cyclization of the resulting products into lactones 1 (Scheme 2). It should be noted that this method employs reagents with their usual polarities, with the configuration of the C-4 stereocenter of the target product being set at the stage of the C–C bond forming reaction.
Thus, the suggested strategy starts with the coupling of nitronates 2 with silyl ketene acetals. This transformation, like other Mukaiyama–Mannich reactions, requires a Lewis acid catalyst – tert-butyldimethylsilyl triflate (TBSOTf), in particular – for the production of nitroso acetals 3 (Scheme 2) in a rather highly diastereoselective fashion.13,14 The products 3 contain the complete carbon skeleton of the target amines 1 and for their transformation into lactones 1, only the transformations of the functional groups is required. However, the direct reduction of compounds 3 into amines 1 with both N–O bonds cleavage and lactone ring closure, within a one step protocol turned out to be ineffective.13 A more convenient approach seems to employ two steps (Scheme 2): (1) lactone cycle formation via oxidation of isoxazolidines 3 into nitro compounds 4; (2) selective nitro group reduction in lactones 4. This modified scheme is supported with our recent research on the six-membered analogues of nitroso acetals 3 which are effectively oxidized with mCPBA into the corresponding nitro compounds with the retention of the relative configuration of all the stereocenters.15,16
 | ||
| Scheme 2 Three-step protocol for synthesis of the target amines 1 via intermediacy of nitro lactones 4. | ||
A nitro group connected to a tertiary carbon in lactones 4 can be a priori regarded as a latent amine function, due its inertness to common reagents.17 Therefore, in this manuscript, the main accent was on the preparation of the nitroso acetals 3 (step 2 → 3) and their oxidation to nitro lactones 4.
Results and discussion
Starting from the available nitronates 2 (ref. 11), a representative series of functionalized nitroso acetals 3a–h was obtained via silyl ketene acetal addition, according to a well-known protocol,13 or its modified version (Table 1, see the Experimental section for details). Only three of these: compounds trans-3a, 3g (inseparable mixture of isomers trans/cis 5.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and trans-3d (containing 8% of the cis-isomer) have been obtained previously.13 The synthesis of the derivatives trans-3b and cis-3b proceeded in a rather similar way, although for 3e the trans-selectivity was apparently decreased in comparison with the analogous 3d.
:1) and trans-3d (containing 8% of the cis-isomer) have been obtained previously.13 The synthesis of the derivatives trans-3b and cis-3b proceeded in a rather similar way, although for 3e the trans-selectivity was apparently decreased in comparison with the analogous 3d.

To the best of our knowledge, no 4-substituted nitronates 2 were introduced in this type of reaction beforehand. These compounds were found to exhibit nearly a 100% trans-selectivity in coupling with the silyl ketene acetal, but required prolonged reaction times. The synthesis of the 4-phenyl-substituted trans-3c was completed in only 24 h with 0.2 equivalents of TBSOTf. The synthesis of the even more bulky bicyclic trans-3f required a full equivalent of the promoter within 8 h exposure at −78 °C. The trans-position of the substituent at C-4 and the introduced ketene acetal residue was proved by NOE NMR experiments (see Fig. 3).
 | ||
| Fig. 2 Molecular structures of 4b and 4h presented in thermal ellipsoids at 50% probability. Hydrogen atoms are omitted for clarity. | ||
 | ||
| Fig. 3 Key NOE correlations for trans-3c and trans-3f. | ||
The electrophilic activity of the nitronate 2h, containing a halomethyl group at C-3, might appear in two different fashions – nucleophile addition at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, or its halogenation.18 Fortunately, the nitronate 2h (R′ = Br) reacted with the silyl ketene acetal in a usual fashion, to give the respective nitroso acetal 3h (trans/cis = 8:1).
N bond, or its halogenation.18 Fortunately, the nitronate 2h (R′ = Br) reacted with the silyl ketene acetal in a usual fashion, to give the respective nitroso acetal 3h (trans/cis = 8:1).
For the elaboration of the second stage of the suggested protocol (Scheme 2, 3 → 4) the oxidation was optimized on the model nitroso acetal 3a. When the conditions, previously reported for 6-membered cyclic analogues of 3,15 were applied to 3a (Scheme 3), protocol (i), the target nitro lactone 4a was obtained in only a 55% yield, along with the nitrous acid elimination product 5a (8%) and a mixture of unidentified linear by-products. The best result (Scheme 3), conditions (ii), achieved with sole mCPBA utilization and prolongation of the reaction time up to 3 days, accomplished the lactone ring-closure step. With the latter process in hand, this increased the isolated yield of the nitro lactone 4a to 85%.
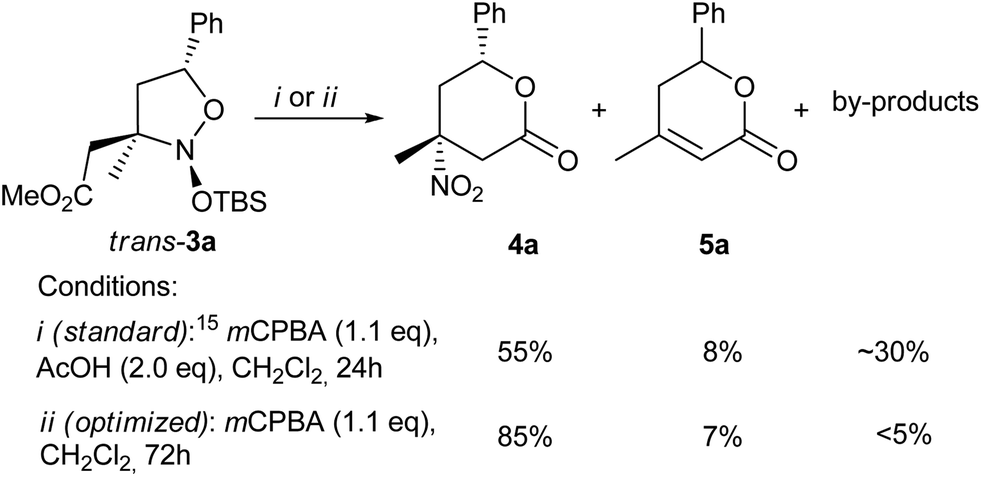 | ||
| Scheme 3 Optimization of nitroso acetal 3 oxidation. | ||
The optimized oxidation conditions were applied to the whole nitroso acetal 3 series. The individual diastereomers of the 5- or 4-aryl substituted nitroso acetals trans-3a–c and cis-3b were smoothly oxidized to give the desired lactones 4a–c and 4b′ in moderate to high yields with the retention of configuration of all the stereocenters (Table 2, entries 1–4). The R′CH2-substituted at C-3 nitroso acetals 3f–h (R′ = Ph, Br, entries 7–9), that were usually used as diastereomeric mixtures, also gave the target nitro lactones 4f–h, but a longer reaction time was required. When the nitroso acetals 3d,e, with the electron-withdrawing CO2Me group at C-5 were employed (entries 5–6), only the formation of acyclic γ-nitro alcohols 6d,e – the proposed precursors of lactones 4 – was observed, reasonably due to the lower nucleophilicity of the alcohol function in these substrates. Even more acidic conditions (Amberlyst-15®), did not manage to achieve the cyclization of 6d and 6e into the corresponding lactones, 4d and 4e.
|
|||
|---|---|---|---|
| # | Nitroso acetal 3 (substituents) | Reaction time, days | Product, yield, % (trans/cis dra) |
| a Determined by 1H NMR spectra. | |||
| 1 | trans-3a | 3 | 4a, 85 |
| (R = 5-Ph, R′ = H) | |||
| 2 | trans-3b | 3 | 4b, 79 |
| (R = 5-(4-BrC6H4), R′ = H) | |||
| 3 | cis-3b | 3 | 4b′, 63 |
| (R = 5-(4-BrC6H4), R′ = H) | |||
| 4 | trans-3c | 2 | 4c, 76 |
| (R = 4-Ph, R′ = H) | |||
| 5 | 3d, trans/cis, 11/1 | 1 | 6d/6d′, 95 (15:1) |
| (R = 5-CO2Me, R′ = H) | |||
| 6 | 3e, trans/cis, 5.2/1 | 1 | 6e/6e′, 92 (6.3:1) |
| (R = 5-CO2Me and 5-Me, R′ = H) | |||
| 7 | trans-3f | 7 | 4f, 64 |
| (R = cis-4,5-(CH2)3-, R′ = Ph) | |||
| 8 | 3g, trans/cis, 5.5/1 | 5 | 4g/4g′, 79 (6.5:1) |
| (R = 5-Ph, R′ = Ph) | |||
| 9 | 3h, trans/cis, 8/1 | 5 | 4h, 85 (>20:1) |
| (R = 5-Ph, R′ = Br) | |||

The configurations of compounds 4b and 4h were supported by X-ray diffraction analysis (Fig. 2). The stereochemistry of the other nitro lactones 4 was assigned by the similarity of their coupling constants with those of 4b and 4h in the 1H NMR spectra. The nitro group formation in all of the compounds 4 was proven by 14N NMR.
The last step of the lactones 1 preparation – selective reduction of the nitro group – despite many precedents in the literature for tertiary nitro compounds17,19 – turned out to be rather capricious. The standard hydrogenation conditions, like Pd/C in MeOH performed on 4a gave only the acyclic product 7 in almost a quantitative yield as the result of the hydrogenolysis of the CH(Ph)–OC(O) fragment (Scheme 4). The other screened reagents (NaBH4/NiCl2,20 Fe/AcOH,19 Fe/NH4Cl19 and even H2/NiRa17,19) gave mainly the “HNO2” elimination product 5a, along with by-products, of unknown structures. [A more convenient protocol for the synthesis of enones 5 from lactones 4 was developed with the utilization t-BuOK in THF. It furnished compounds 5a and 5c with 94% and 95% yields, within 15–30 minutes, respectively (Scheme 4)].
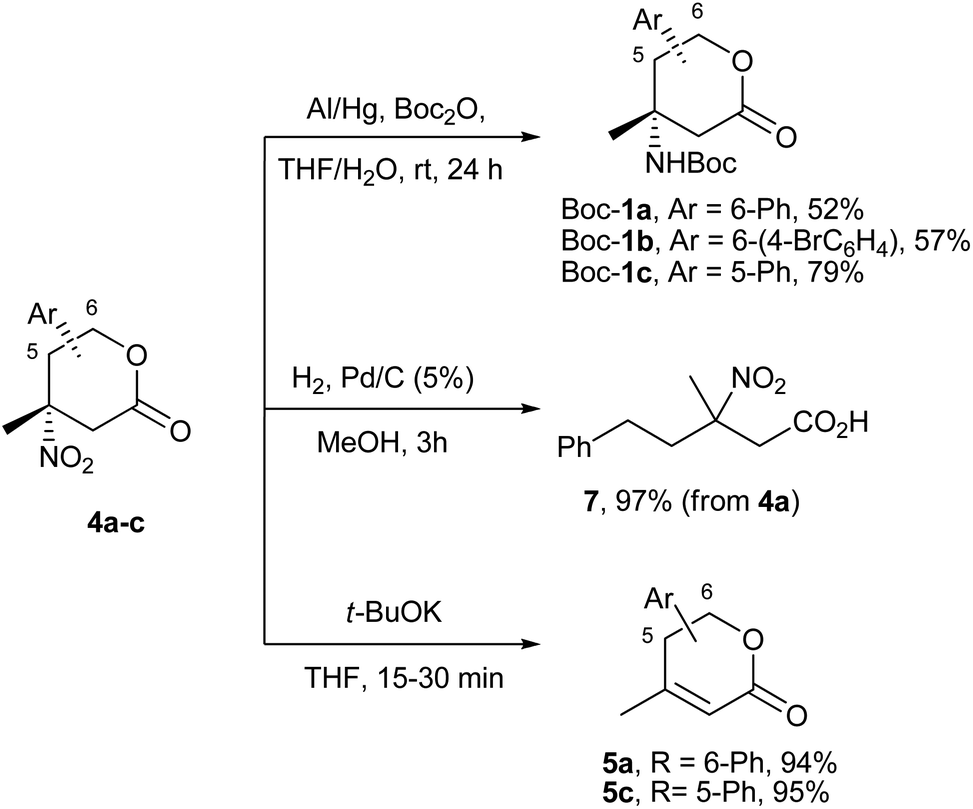 | ||
| Scheme 4 Reduction and accompanying reactions of nitro lactones 4a–c. | ||
In these circumstances, we managed to reduce the nitro group with the rarely used amalgamated aluminium.21 The most complicated intermediates – nitro compounds 4a and 4b with the benzyl carboxylate function – gave the protected derivatives Boc-1a and Boc-1b of target amines in 52 and 57% yields, respectively (Scheme 4). For the extremely sterically hindered lactone 4c, a slightly modified procedure was applied, when the reduction and protection were divided into two technical steps. By this methodology and the utilization of a catalytic amount of DMAP for the Boc2O activation, the desired product Boc-1c was obtained in a 79% yield.
Conclusions
In summary, we have demonstrated that the target β-amino-δ-lactones 1, or more precisely, their N-Boc derivatives, can be obtained from the five-membered cyclic nitronates 2, according to the suggested three-step protocol. The novelty of the approach consists of the combination of three transformations (C,C-coupling, oxidation and selective reduction of the NO2 group). This procedure was applied to the synthesis of scaffolds 1 for the first time. In our opinion, there are no obstacles for the accomplishment of this strategy in an asymmetric manner.Experimental section
General remarks
Reactions with TBSOTf were performed in oven-dried (150 °C) glassware under an argon atmosphere. The NMR spectra were recorded on a Bruker AM-300 (1H: 300.13 MHz, 13C: 75.47 MHz, 14N: 21.69 MHz, 29Si: 59.63 MHz) and Bruker AMX-400 (1H: 400.1 MHz, 13C: 100.6 MHz) and referenced to a residual solvent peak. The chemical shifts are reported in ppm (δ); multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet) and br (broad). The ratios of the stereoisomers were derived from the relative integral intensities of the characteristic signals in the 1H NMR spectra. Coupling constants, J, are reported in Hertz. Key NOESY correlations are shown with arrows (Fig. 2). The IR spectra were recorded on a Bruker VEKTOR-22 in the range 400–4000 cm−1 (resolution 2 cm−1) as a thin layer. The melting points were determined on Kofler melting point apparatus and are uncorrected. The elemental analyses were performed by the Analytical Laboratory of the N. D. Zelinsky Institute of Organic Chemistry. The HR mass spectra were recorded on a Bruker MicroTOFF spectrometer with electrospray ionization (ES-I).The X-ray diffraction measurements were carried out using SMART 1000 CCD and Smart APEX II diffractometers at 100 K. The frames were integrated and corrected for absorption by the APEX 2 program package [APEX2 Software Package, Bruker AXS Inc., 5465, East Cheryl Parkway, Madison, WI 5317, 2005]. The details of crystallographic data and experimental conditions are given in Table SI1 in the ESI.† The structures were solved by a direct method and refined by the full-matrix least-squares technique against F2 in the anisotropic–isotropic approximation. The hydrogen atoms were located from the difference Fourier maps and refined in a rigid body model. All the calculations were performed using the APEX 2 program package [APEX2 Software Package, Bruker AXS Inc., 5465, East Cheryl Parkway, Madison, WI 5317, 2005].
The analytical thin-layer chromatography was performed on silica gel plates, with the QF-254 indicator. The visualization of the TLC plates was accomplished with a UV light and/or anisaldehyde/H2SO4. The preparative liquid chromatography was performed on columns with “Merck”-silica (Kieselgel 60, 230–400 mesh). All the solvents for the chromatography and extractions were of technical grade and distilled prior to use. The following solvents and reagents were distilled from the indicated drying agents: CH2Cl2, CHCl3, Et3N (CaH2), MeOH (Mg), THF, dioxane (LiAlH4).
Synthesis of nitroso acetals 3 (first step, see Scheme 3)
:1).C19H30NO4BrSi (444.44): calcd C, 51.35; H, 6.80; N, 3.15; found C, 51.66; H, 7.05; N, 3.38%.
:1 (inseparable mixture), colorless oil; TLC: Rf = 0.66 (hexane–EtOAc, 1/1); 1H NMR (400.1 MHz, 305 K, CDCl3): rel-2S,3R,5R-isomer, trans-3e (major): δ = 0.11 and 0.20 [2s, 6H, Si(CH3)2], 0.89 [s, 9H, t-Bu], 1.13 [s, 3H, NCCH3], 1.69 [s, 3H, OCCH3], 2.50 [d, 2J = 14.7 Hz, 1H, CHAHB], 2.51 [d, 2J = 12.8 Hz, 1H, CHCHD(CO2Me)], 2.82 [d, 2J = 14.7 Hz, 1H, CHAHB], 2.85 [d, 2J = 12.8 Hz, 1H, CHCHD(CO2Me)], 3.66 [s, 3H, CO2CH3], 3.73 [s, 3H, CH2CO2CH3]; rel-2R,3S,5R-isomer, cis-3e (minor): δ = 0.08 and 0.17 [2s, 6H, Si(CH3)2], 0.84 [s, 9H, t-Bu], 1.27 [s, 3H, NCCH3], 1.53 [s, 3H, OCCH3], 2.50 [d, 2J = 14.7 Hz, 1H, CHAHB], 2.51 [d, 2J = 12.8 Hz, 1H, CHCHD(CO2Me)], 2.82 [d, 2J = 14.7 Hz, 1H, CHAHB], 2.85 [d, 2J = 12.8 Hz, 1H, CHCHD(CO2Me)], 3.66 [s, 3H, CH2CO2CH3], 3.68 [s, 3H, CO2CH3] ppm; 13C NMR (100.6 MHz, 305 K, CDCl3): rel-2S,3R,5R-isomer, trans-3e (major): δ = −5.0 and −4.4 [Si(CH3)2], 17.8 [C(CH3)3], 22.8 [NCCH3], 26.8 [C(CH3)3], 28.5 [OCCH3], 42.1 [CH2CO2Me], 46.5 [CH2], 51.7 [CH2CO2CH3], 52.6 [CO2CH3], 74.4 [N–C], 88.6 [N–O–C], 171.4 [CH2CO2Me], 174.0 [CO2Me]; rel-2R,3S,5R-isomer, cis-3e (minor): δ = −4.9 and −4.4 [Si(CH3)2], 17.9 [C(CH3)3], 24.3 [NCCH3], 25.2 [C(CH3)3], 25.8 [OCCH3], 42.3 [CH2CO2Me], 44.1 [CH2], 51.7 [CH2CO2CH3], 52.3 [CO2CH3], 74.8 [N–C], 86.4 [N–O–C], 171.5 [CH2CO2Me], 175.1 [CO2Me] ppm. 29Si NMR (305 K, CDCl3, INEPT): rel-2S,3R,5R-isomer, trans-3e (major): δ = 26.0; rel-2R,3S,5R-isomer, cis-3e (minor): δ = 26.2 ppm. HRMS (ES-I) calcd for C16H31NO6SiH [M + H]+ 362.1993; found 362.1990.:1, slightly greenish oil, TLC: Rf = 0.85 (hexane–EtOAc, 1/1); 1H NMR (300.13 MHz, 300 K, CDCl3): δ = −0.21 and 0.09 [2s, 6H, Si(CH3)2], 0.87 [s, 9H, t-Bu], 1.66–1.80 [m, 4H, CH2Cyc.], 1.90–2.00 and 2.11–2.21 [2m, 2H, CH2Cyc.] 2.27 [d, 2J = 16.3 Hz, 1H, CHAHB], 2.73 [d, 2J = 16.3 Hz, 1H, CHAHB], 2.83 [d, 2J = 13.7 Hz, 1H, CHCHDPh], 3.02 [d, 2J = 13.7 Hz, 1H, CHCHDPh], 3.06 [t, 3J ≈ 3J = 7.3 Hz, 1H, CH], 3.71 [s, 3H, CO2Me], 5.16 [dd, 3J = 6.4, 3J = 5.3 Hz, 1H, OCH], 7.16–7.31 [m, 5H, Ph] ppm. 13C NMR (75.47 MHz, 300 K, CDCl3): δ = −5.2 and −5.0 [Si(CH3)2], 17.8 [C(CH3)3], 25.9 [C(CH3)3], 26.6, 27.9, 30.6, 37.4 and 38.9 [CH2], 51.4 [OCH3], 53.3 [CH], 79.0 [C–N], 87.4 [OCH], 126.2 [CHp-Ph], 127.5 and 128.5 [CHo-Ph and CHm-Ph], 137.9 [Ci-Ph], 172.0 [OC = O] ppm; 29Si NMR (300 K, CDCl3): δ = 25.1 ppm; HRMS (ES-I) calcd for C22H35NO4SiNa [M + Na]+ 428.2228; found 428.2208.:1 (inseparable mixture), TLC: Rf = 0.76 (hexane–EtOAc, 1/1); 1H NMR (300.13 MHz, 302 K, CDCl3): rel-2S,3R,5R-isomer, trans-3h (major): δ = 0.22 and 0.26 [2s, 6H, Si(CH3)2], 0.97 [s, 9H, t-Bu], 2.65 [dd, 2J = 13.2, 3J = 10.2 Hz, 1H, CHAHB], 2.92 [dd, 2J = 13.2, 3J = 8.8 Hz, 1H, CHAHB], 3.00 [d, 2J = 16.9 Hz, 1H, CHCHD(CO2Me)], 3.12 [d, 2J = 16.9 Hz, 1H, CHCHD(CO2Me)], 3.64 [d, 2J = 11.0 Hz, 1H, CHEHFBr], 3.72 [s, 3H, CO2Me], 3.78 [d, 2J = 11.0 Hz, 1H, CHEHFBr], 5.63 [dd, 3J = 8.8, 3J = 6.6, 1H, CH], 7.28–7.48 [m, 5H, Ph]; rel-2R,3S,5R-isomer, cis-3h (minor): δ = 0.04 and 0.20 [2s, 6H, Si(CH3)2], 0.90 [s, 9H, t-Bu], 2.32 [dd, 2J = 13.2, 3J = 6.6 Hz, 1H, CHAHB], 2.87–3.01 [m, 2H, CHAHB and CHCHD(CO2Me), overlaps with major isomer], 3.14 [d, 2J = 16.9 Hz, 1H, CHCHD(CO2Me)], 3.67 [d, 2J = 17.6 Hz, 1H, CHEHFBr], 3.72 [s, 3H, CO2Me], 3.78 [d, 2J = 17.6 Hz, 1H, CHEHFBr], 5.63 [dd, 3J = 10.2, 3J = 8.1 Hz, 1H, CH], 7.28–7.48 [m, 5H, Ph] ppm; 13C NMR (75.47 MHz, 302 K, CDCl3): rel-2S,3R,5R-isomer, trans-3h (major): δ = −5.2 and −4.8 [Si(CH3)2], 18.0 [C(CH3)3], 25.9 [C(CH3)3], 37.8 and 37.9 [CH2CO2Me and CH2Br], 42.3 [CH2], 51.7 [OCH3], 77.3 [C–N], 80.3 [OCH], 126.4 [CHp-Ph], 128.0 and 128.7 [CHo-Ph and CHm-Ph], 139.1 [Ci-Ph], 171.0 [OC = O]; rel-2R,3S,5R-isomer, cis-3h (minor): δ = −4.9 and −4.5 [Si(CH3)2], 17.8 [C(CH3)3], 25.7 [C(CH3)3], 35.1 [CH2Br], 37.4 [CH2CO2Me], 41.8 [CH2], 51.7 [OCH3], 76.5 [C–N], 84.7 [OCH], 127.3, 127.7 and 128.2 [CHPh], 140.1 [Ci-Ph], 170.8 [OC = O] ppm; HRMS (ES-I) calcd for C19H30BrNO4SiNa [M + Na]+ 466.1020 and 466.1000; found 466.1023 and 466.1002.Nitro lactone 4 formation (second step, see Scheme 3)
O), 1547 (vs., νasNO2), 1354 (s, νsNO2); 1H NMR (300.13 MHz, 299 K, CDCl3): δ = 1.87 [s, 3H, CH3], 2.56 [dd, 2J = 15.5, 3J = 3.7 Hz, 1H, CHAHB], 2.63 [dd, 2J = 15.5, 3J = 11.4 Hz, 1H, CHAHB], 2.98 [d, 2J = 16.9 Hz, 1H, CHCHD], 3.35 [d, 2J = 16.9 Hz, 1H, CHCHD)], 5.32 [dd, 3J = 11.4, 3J = 3.7 Hz, 1H, CH], 7.33–7.45 [m, 5H, Ph] ppm; 13C NMR (75.47 MHz, 299 K, CDCl3): δ = 25.8 [CH3], 40.0 [CH2], 42.0 [CH2CO], 77.2 [CH], 84.4 [CNO2], 126.0 and 128.6 [CHo-Ph and m-Ph], 129.1[(CHp-Ph], 137.2 [Ci-Ph], 167.6 [OCO] ppm; 14N NMR (299 K, CDCl3): δ = 16 (ν1/2 ≈ 100 Hz) ppm. C12H13NO4 (235.24): calcd C, 61.27; H, 5.57; N, 5.95; found C, 61.36; H, 5.72; N, 5.85%.O] ppm; 14N NMR (312 K, CDCl3): δ = 13 (ν1/2 ≈ 150 Hz) ppm; C12H12BrNO4 (314.13): calcd C, 45.88; H, 3.85; N, 4.46; found C, 45.62; H, 3.77; N, 4.21%.O], 48.4 [CH], 68.5 [OCH2], 87.7 [CNO2], 128.5 and 129.2 [CHo-Ph and CHm-Ph], 129.4 [CHp-Ph], 132.4 [Ci-Ph], 166.7 [OCO] ppm; 14N NMR (28.9 MHz, 299 K, CDCl3): δ = 7 (ν1/2 ≈ 100 Hz) ppm; C12H13NO4 (235.24): calcd C, 61.27; H, 5.57; N, 5.95; found C, 60.99; H, 5.56; N, 5.96%.O] ppm; 14N NMR (299 K, CDCl3): δ = 14 (ν1/2 ≈ 200 Hz) ppm; HRMS (ES-I) calcd for C9H15NO7Na [M + Na]+ 272.0741; found 272.0747.:1 (inseparable mixture), oil, TLC: Rf = 0.32 (hexane–EtOAc, 1/1); 1H NMR (300.13 MHz, 299 K, CDCl3): rel-2R,4R-isomer 6e (major): δ = 1.41 [s, 3H, CH3], 1.68 [s, 3H, CH3], 2.67 [s, 2H, CH2], 2.95 [d, 2J = 16.4 Hz, 1H, CHAHB], 3.09 [d, 2J = 16.4 Hz, 1H, CHAHB], 3.20 [br s, 1H, OH], 3.68 [s, 3H, CO2Me], 3.79 [s, 3H, CO2Me]; rel-4S,6R-isomer 6e′ (minor): δ = 1.42 [s, 3H, CH3], 1.64 [s, 3H, CH3], 2.64 [d, 2J = 15.2 Hz, 1H, CHCHD], 2.74 [d, 2J = 15.2 Hz, 1H, CHCHD], 3.12 [d, 2J = 17.2 Hz, 1H, CHAHB], 3.20 [br s, 1H, OH], 3.25 [d, 2J = 17.2 Hz, 1H, CHAHB], 3.68 [s, 3H, CO2Me], 3.80 [s, 3H, CO2Me] ppm; 13C NMR (75.47 MHz, 299 K, CDCl3): rel-4R,6R-isomer 6e (major): δ = 23.0 and 29.7 [CH3], 43.1 and 45.8 [CH2], 52.0 and 53.2 [OCH3], 73.0 [C], 86.6 [CNO2], 169.4 and 176.9 [OCO]; rel-4S,6R-isomer 6e′ (minor): δ = 23.9 and 29.6 [CH3], 41.9 and 45.3 [CH2], 51.9 and 53.2 [OCH3], 72.9 [C], 87.4 [CNO2], 169.9 and 176.9 [OCO] ppm; 14N NMR (299 K, CDCl3): δ = 19 (ν1/2, ca. 200 Hz) ppm; HRMS (ES-I) calcd for C10H17NO7Na [M + Na]+ 286.0897; found 286.0900.O] ppm; 14N NMR (299 K, CDCl3): δ = 8 (ν ½ ≈ 230 Hz) ppm; HRMS (ES-I) calcd for C15H17NO4Na [M + Na]+ 298.1050; found 298.1050.:1 (inseparable mixture), mp = 129–135 °C (Et2O), TLC: Rf = 0.47 (hexane–EtOAc, 1/1); 1H NMR (300.13 MHz, CDCl3): rel-4R,6R-isomer 4g (major): δ = 2.65 [dd, 2J = 15.4, 3J = 11.7 Hz, 1H, CHAHB], 2.78 [dd, 2J = 15.4, 3J = 3.7 Hz, 1H, CHAHB], 3.07 [d, 2J = 16.9 Hz, 1H, CHCHD], 3.26 [d, 2J = 16.9 Hz, 1H, CHCHD], 3.41 [d, 2J = 14.0 Hz, 1H, CHEHF], 3.52 [d, 2J = 14.0 Hz, 1H, CHEHF], 5.12 [dd, 3J = 11.7, 3J = 3.7 Hz, 1H, CH], 7.11–7.19 [m, 2H, CHPh], 7.25–7.44 [m, 8H, Ph]; rel-4S,6R-isomer 4g′ (minor): δ (selected signals) = 2.86 [d, 2J = 14.0 Hz, 1H, CHCHD], 3.50 [d, 2J = 14.0 Hz, 1H, CHEHF], 5.22 [dd, 3J = 12.5, 3J = 2.0 Hz, 1H, CH] ppm; 13C NMR (75.47 MHz, CDCl3): rel-4R,6R-isomer 4g (major): δ = 37.6, 40.3, 44.8, [3 CH2], 77.0 [CH], 87.9 [CNO2], 125.8, 128.6, 129.0, 129.1, 129.2 and 129.9 [CHPh], 132.4 and 137.5 [Ci-Ph], 167.4 [OC = O], rel-4S,6R-isomer 4g′ (minor): δ = 37.1, 40.4 and 45.8 [3 CH2], 77.3 [CH], 87.9 [CNO2], 126.0, 127.3, 128.4, 128.9, 129.0 and 129.9 [CHPh], 132.0 and 138.1 [Ci-Ph], 166.7 [OCO] ppm; 14N NMR (299 K, CDCl3): δ = 15 (ν 1/2, ca. 200 Hz) ppm; HRMS (ES-I) calcd for C18H17NO4Na [M + Na]+ 334.1050; found 334.1050.:1, mp = 92–95 °C (Et2O), TLC: Rf = 0.43 (hexane–EtOAc, 1/1); 1H NMR (300.13 MHz, 299 K, CDCl3): δ = 2.70–2.84 [m, 2H, CH2], 3.20 [d, 2J = 16.9 Hz, 1H, CHAHB], 3.41 [d, 2J = 16.9 Hz, 1H, CHAHB], 3.95–4.09 [m, 2H, CH2Br], 5.32 [dd, 3J = 9.5, 3J = 5.2 Hz, 1H, CH], 7.32–7.49 [m, 5H, Ph] ppm; 13C NMR (75.47 MHz, 299 K, CDCl3): δ = 35.2 [CH2Br], 37.2 and 40.1 [CH2 and CH2CO], 77.1 [CH], 86.4 [CNO2], 125.9 and 129.0 [CHo-Ph and CHm-Ph], 129.3 [CHp-Ph], 136.4 [Ci-Ph], 168.6 [OCO] ppm; 14N NMR (21.69 MHz, 299 K, CDCl3): δ = 6 (ν1/2, ca. 170 Hz) ppm; C12H12BrNO4 (314.13): calcd C, 45.88; H, 3.85; N, 4.46; found: C, 46.00; H, 3.87; N, 4.27%.Reduction of nitro lactones 4 into carbamates Boc-1 (third step, see Scheme 3)
OBoc], 170.3 [OC = O] ppm; HRMS (ES-I) calcd for C17H23NO4Na [M + Na]+ 328.1519; found 328.1513.OBoc], 169.7 [OCO] ppm; HRMS (ES-I) calcd for C17H22BrNO4Na [M + Na]+ 406.0624; found 406.0619.OBoc], 176.9 [OCO] ppm; HRMS (ES-I) calcd for C17H23NO4Na [M + Na]+ 328.1519; found 328.1523.Other transformations of nitro lactones 4
CH], 126.1, 128.6 and 128.7 [CHPh], 138.7 [Ci-Ph], 157.0 [C], 164.9 [OCO] ppm.CH], 128.0 [CHp-Ph], 128.1 and 129.1 [CHPh], 137.1 [Ci-Ph], 159.2 [C], 164.2 [OCO]; HRMS (ES-I) calcd for C12H12O2 [M + Na]+ 211.0730; found 211.0731.X-Ray data
Single crystals, suitable for X-ray diffraction analysis, were obtained by the slow cooling and slow evaporation of CHCl3 solutions of 4b, 4h, saturated at reflux.Acknowledgements
This work was performed with the financial support of the Russian Foundation for Basic Research (Grant # 12-03-00278-a) and the President of Russian Federation Program for supporting young scientists and leading scientific schools (Grant NSh-412.2012.3).Notes and references
- T. Taga, N. Akimoto and T. Ibuka, Chem. Pharm. Bull., 1984, 32(10), 4223–4225 CrossRef CAS.
- Y. Yang, W.-J. Zuo, Y.-X. Zhao, W.-H. Dong, W.-L. Mei and H.-F. Dai, Planta Med., 2012, 78, 1881–1884 CrossRef CAS PubMed.
- (a) K. Tsuda, S. Ikuma, M. Kawamura, R. Tachikawa, K. Sakai, C. Tamura and O. Amalasu, Chem. Pharm. Bull., 1964, 12, 1357–1374 CrossRef CAS; (b) R. B. Woodward, Pure Appl. Chem., 1964, 9, 49–74 CrossRef CAS; (c) T. Goto, Y. Kishi, S. Takahashi and Y. Hirata, Tetrahedron, 1965, 21, 2059–2088 CrossRef CAS.
- (a) M. Yotsu-Yamashita, Y. Yamagishi and T. Yasumoto, Tetrahedron Lett., 1995, 36, 9329–9332 CrossRef CAS; (b) T. Nakagawa, J. Jang and M. Yotsu-Yamashita, Anal. Biochem., 2006, 352, 142–144 CrossRef CAS PubMed.
- Tetrodotoxin, Saxitoxin, and the Molecular Biology of the Sodium Channel, ed. C. Y. Kao and S. R. Levinson, New York Academy of Sciences, New York, 1986, vol. 479 Search PubMed.
- U. Koert, Angew. Chem., Int. Ed., 2004, 43, 5572–5576 CrossRef CAS PubMed.
- (a) Y. Kishi, F. Nakatsubo, M. Aratani, T. Goto, S. Inoue, H. Kakoi and S. Sugiura, Tetrahedron Lett., 1970, 11, 5127–5128 CrossRef; (b) Y. Kishi, F. Nakatsubo, M. Aratani, T. Goto, S. Inoue and H. Kakoi, Tetrahedron Lett., 1970, 11, 5129–5132 CrossRef; (c) Y. Kishi, M. Aratani, T. Fukuyama, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura and H. Kakoi, J. Am. Chem. Soc., 1972, 94, 9217–9219 CrossRef CAS; (d) Y. Kishi, T. Fukuyama, M. Aratani, F. Nakatsubo, T. Goto, S. Inoue, H. Tanino, S. Sugiura and H. Kakoi, J. Am. Chem. Soc., 1972, 94, 9219–9221 CrossRef CAS.
- (a) N. Ohyabu, T. Nishikawa and M. Isobe, J. Am. Chem. Soc., 2003, 125, 8798–8805 CrossRef CAS PubMed; (b) T. Nishikawa, D. Urabe and M. Isobe, Angew. Chem., Int. Ed., 2004, 43, 4782–4785 CrossRef CAS PubMed.
- (a) T. Umezawa, T. Hayashi, H. Sakai, H. Teramoto, T. Yoshikawa, M. Izumida, Y. Tamatani, T. Hirose, Y. Ohfune and T. Shinada, Org. Lett., 2006, 8, 4971–4974 CrossRef CAS PubMed; (b) T. Umezawa, T. Shinada and Y. Ohfune, Chem. Lett., 2010, 39, 1281–1282 CrossRef CAS.
- A. Hinman and J. Du Bois, J. Am. Chem. Soc., 2003, 125, 11510–11511 CrossRef CAS PubMed.
- S. L. Ioffe, in Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis, ed. H. Feuer, John Wiley & Sons, Chichester, United Kingdom, 2nd edn, 2008, pp. 435–748 Search PubMed.
- S. Saaby, K. Nakama, M. A. Lie, R. G. Hazell and K. A. Jørgensen, Chem.–Eur. J., 2003, 9, 6145–6154 CrossRef CAS PubMed.
- V. O. Smirnov, A. S. Sidorenkov, Yu. A. Khomutova, S. L. Ioffe and V. A. Tartakovsky, Eur. J. Org. Chem., 2009, 3066–3074 CrossRef CAS.
- About C-C- and C-N-coupling of other nitronate types with various nucleophiles, see (a) V. O. Smirnov, S. L. Ioffe, A. A. Tishkov, Yu. A. Khomutova, I. D. Nesterov, M. Y. Antipin, W. A. Smit and V. A. Tartakovsky, J. Org. Chem., 2004, 69, 8485–8488 CrossRef CAS PubMed; (b) Yu. A. Khomutova, V. O. Smirnov, H. Mayr and S. L. Ioffe, J. Org. Chem., 2007, 72, 9134–9140 CrossRef CAS PubMed; (c) V. O. Smirnov, Yu. A. Khomutova, V. A. Tartakovsky and S. L. Ioffe, Eur. J. Org. Chem., 2012, 3377–3384 CrossRef CAS; (d) A. A. Mikhaylov, A. D. Dilman, Yu. A. Khomutova, D. E. Arkhipov, A. A. Korlyukov and S. L. Ioffe, Eur. J. Org. Chem., 2013, 5670–5677 CrossRef CAS.
- A. S. Naumova, A. A. Mikhaylov, M. I. Struchkova, Yu. A. Khomutova, V. A. Tartakovsky and S. L. Ioffe, Eur. J. Org. Chem., 2012, 2219–2224 CrossRef CAS.
- The other only example on the oxidation of N-silyloxy isoxazolines with mCPBA described in literature, see (a) V. F. Rudchenko, I. I. Chervin, A. E. Aliev and R. G. Kostyanovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci. (Engl. Transl.), 1991, 40, 1380–1386 CrossRef (Russian: Izv. Akad. Nauk SSSR, Ser. Khim., 1991, 1563–1568); oxidation with unspecified oxidant upon TBAF treatment; (b) P.-Y. Roger, A.-C. Durand, J. Rodriguez and J.-P. Dulcère, Org. Lett., 2004, 6, 2027–2029 CrossRef CAS PubMed; (c) E. Dumez, A.-C. Durand, M. Guillaume, P.-Y. Roger, R. Faure, J.-M. Pons, G. Herbette, J.-P. Dulcère, D. Bonne and J. Rodriguez, Chem.–Eur. J., 2009, 15, 12470–12488 CrossRef CAS PubMed.
- N. Ono, The Nitro Group in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2001, p. 372 Search PubMed.
- A. A. Mikhaylov, A. D. Dilman, R. A. Kunetsky, Yu. A. Khomutova, M. I. Struchkova, A. A. Korlyukov, S. L. Ioffe and V. A. Tartakovsky, Tetrahedron Lett., 2010, 51, 1038–1040 CrossRef CAS PubMed.
- S. L. Ioffe, V. A. Tartakovsky and S. S. Novikov, Russ. Chem. Rev., 1966, 35, 19–32 CrossRef PubMed.
- J. O. Osby and B. Ganem, Tetrahedron Lett., 1985, 26, 6413–6416 CrossRef CAS.
- (a) R. W. Fitch and F. A. Luzzio, Tetrahedron Lett., 1994, 35, 6013–6016 CrossRef CAS; (b) J. C. Anderson and H. A. Chapman, Synthesis, 2006, 3309–3315 CrossRef CAS PubMed.
- E. J. Corey and M. Chaykovsky, J. Am. Chem. Soc., 1965, 87, 1345–1353 CrossRef CAS.
- R. W. Dugger and C. H. Heathcock, J. Org. Chem., 1980, 45, 1181–1185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of 1H, 13C and two-dimensional NMR spectra for all novel compounds. CCDC 959160 and 959161. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47309k |
| This journal is © The Royal Society of Chemistry 2014 |