Chiral transmission to crystal photodimerizations of leucine–methionine quasiracemic assemblies†
Yuhua Lu,
Andrew. J. Bolokowicz,
Sarah A. Reeb,
Joshua D. Wiseman and
Kraig A. Wheeler*
Department of Chemistry, Eastern Illinois University, Charleston, Illinois 61920, USA. E-mail: kawheeler@eiu.edu; Tel: +1 217 581 3119
First published on 14th January 2014
Abstract
We report the deliberate use of molecular shape as a structural tool for generating homochiral photoreactive crystalline materials. Building on the crystal packing tendencies of amino acid quasiracemates to form near inversion related motifs, L-leucine and D-methionine were decorated with sulfonamidecinnamic acid groups. When co-crystallized, the ‘fish hook’ shaped components organize into supramolecular dimers with favorable olefin⋯olefin spacing (3.71 and 4.09 Å). UV exposed samples were processed as single-crystal-to-single-crystal transformations and showed quantitative conversion to the expected enantiopure photodimerization product. Similar crystal motifs and reaction outcomes were achieved using the racemic counterparts suggesting the importance of best-fit scenarios directed by the complementary features of molecular shape and non-bonded contacts.
Recent discoveries and applications of biologically inspired materials provide new engineering solutions of current technological interest to such areas as damage repair, hierarchical structuring, functional adaptation, and self-regulation.1 Given the wealth of structural information retrieved from proteins, it is not surprising that these biomaterials and their structural architectures serve as the entry point for many biomimetic investigations. Their role in transport and signalling is critical to cellular function that can be directly traced to the underlying structural features of the fundamental amino acid components.2 Though the profound effect of these building-blocks to protein form and function begins with their placement along the polypeptide chain, how they recognize and differentiate neighboring residues and motifs is critical. At some level, it is intriguing to consider that protein function persists despite structural modification to the amino acid sequence. Of course each mutation, single point or extensive, incurs a penalty with a magnitude related to the stability of the structural feature.
Systematically interchanging an amino acid residue for one of the other naturally occurring options of the same configuration provides an effective strategy for uncovering a wealth of information of the extent of residue importance (Fig. 1).3 These studies have revealed suitable point mutations for a variety of conditions and systems. Though identifying a site or region as highly mutable or conserved can lead to a more intimate understanding of protein structure and function, this process also draws critical attention to the pairwise commonality of amino acids in the form of index tables.4 These indices reveal the interchange likelihood of pairs of amino acids under various physiological conditions. As a general rule, sets of amino acids that lack commonality such as glycine and tyrosine often provide unlikely partners, while groups with similar structural properties (e.g. hydrophobicity and sterics) such as the valine/isoleucine pair are relatively common.
 | ||
| Fig. 1 Applications of amino acid (AA) interchanges to protein point mutations and small molecule systems. | ||
Conceptually, the notion of amino acid replacement, complete or partial, should also hold true to low molecular weight frameworks. In general, this seems not to be the case and the rarity of such a phenomenon is not unexpected given such substitutions translate to a higher overall enthalpic cost.
If proteinaceous materials represent one end of the structural spectrum, then solitary amino acids anchor the other extreme. To date, reports that combine two amino acids of the same handedness (D/D′ or L/L′) include the co-crystallization of asparagine/aspartic acid5 and threonine/allo-threonine.6 In such cases, solid solutions form with molecular sites populated by a disordered mixture of the two components. Why these sites accommodate both molecules offers insight to tailor-made additives with similar topologies and hydrogen-bond profiles to the host molecule. While a parallel experiment with L-isoleucine and L-allo-isoleucine gave inconclusive results, the same report described 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 co-crystallization of the racemic forms of the components gave crystals with scrambled D and L sites with near equal distribution of the leucine and allo-isoleucine molecules.7 This approach to generating racemic solid solutions has also been applied to serine/threonine.8
:1 co-crystallization of the racemic forms of the components gave crystals with scrambled D and L sites with near equal distribution of the leucine and allo-isoleucine molecules.7 This approach to generating racemic solid solutions has also been applied to serine/threonine.8
Though it has been shown that homochiral and racemic forms of amino acids can tolerate the occlusion of select amino acid additives of the same handedness, this tolerance takes on a distinct sense of recognition when pairs of chemically unique amino acids of opposite chirality combine (e.g. D-Ala and L-Val). With this class of compound, commonly referred to as quasiracemates or quasiracemic mixtures (the term pseudoracemates9 has been reserved for conglomerates constructed from enantiomers), replacement no longer occurs with random distribution, but rather the components organize into well-ordered motifs. Of the more than thirty cases of amino acid-based quasiracemates, all align with the two components related by approximate inversion symmetry.10 This penchant for symmetry draws from the >92% likelihood of ‘true’ racemates to form centrosymmetic motifs11 and highlights the importance of such alignment to close packing and the need for complementary molecular shapes – the same properties that contribute to successful mutations in proteins and the formation of amino acid solid solutions. One additional indication of the importance of near inversion symmetry in the construction of quasiracemic motifs is the ability of these materials to accommodate widely different component topologies, for instance with the L-phenylalanine/D-2-aminobutyric acid10a and L-isoleucine/D-alanine10b systems.
The shared outcome of pairing L/L′ (or D/D′), DL/D′L′, or D/L′ amino acids is the construction of desymmetrized motifs. In the case of racemic solid solutions with near equimolar portions of D/L and D′/L′ components, this asymmetry is largely limited to local motifs, while the impact with L/L′ (or D/D′) can occur at both the global and local environment. Though such studies offer important insight to the structural similarity of amino acids and the factors affecting crystal growth, the inherent disorder of these systems limit their utility to applications that do not require predictable molecular alignment. By contrast, quasiracemic materials are well-ordered with pairs of D and L′ components organized with approximate inversion symmetry. The combination of asymmetry and supramolecular control that defines quasiracemates has attracted considerable attention over the last decade as a viable tool for probing protein structure12 as well as the identification,13 kinetic resolution,14 enantiomeric enrichment15 and polar organization16 of small molecule species.
 | ||
| Scheme 1 | ||
Building on our previous investigations of quasiracemic materials, we recently wondered about the feasibility of merging the structural features imposed by these systems with additional functional properties that could benefit from desymmetrized motifs.17 If such a function were linked to chemical reactivity, e.g. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C groups aligned for photodimerization, then the outcome would be imprinted with the chirality of the original quasiracemate (Fig. 2). Recently we reported the application of the quasiracemic method to solid-state reactions using a D-alanine/L-butyrate sulfonamide system (D-1/L-2) (Scheme 1).17a The complementary features of configuration and molecular topology of the amino acid framework gave rise to desymmetrized hydrogen-bonded dimers that when UV irradiation offered access to single-crystal-to-single-crystal (SCSC)18 enantiocontrolled [2 + 2] photoproducts. Though both supramolecular control19 and enantiopure reaction products20 have been pursued with photodimerization reactions, studies that predictably combine these design features remain rare.17a,21
C groups aligned for photodimerization, then the outcome would be imprinted with the chirality of the original quasiracemate (Fig. 2). Recently we reported the application of the quasiracemic method to solid-state reactions using a D-alanine/L-butyrate sulfonamide system (D-1/L-2) (Scheme 1).17a The complementary features of configuration and molecular topology of the amino acid framework gave rise to desymmetrized hydrogen-bonded dimers that when UV irradiation offered access to single-crystal-to-single-crystal (SCSC)18 enantiocontrolled [2 + 2] photoproducts. Though both supramolecular control19 and enantiopure reaction products20 have been pursued with photodimerization reactions, studies that predictably combine these design features remain rare.17a,21
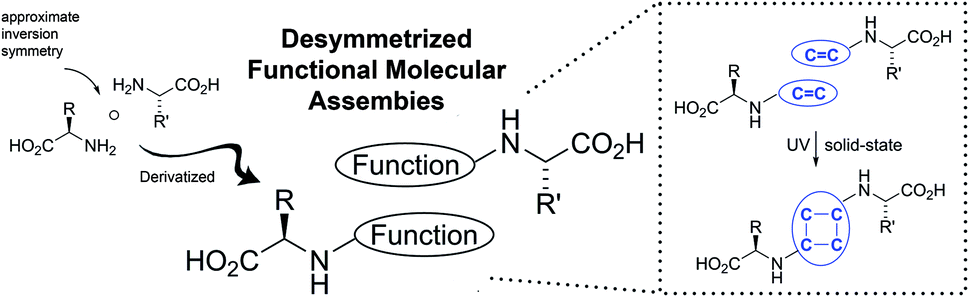 | ||
| Fig. 2 Desymmetrized motifs of amino acid quasiracemates applied to photodimerization reactions. | ||
Given the chiral outcome of UV irradiating crystals of D-1/L-2, the question then arose whether this approach to amplifying stereochemistry in solid-state transformations could be applied to systems decorated with methionine (Met) and leucine (Leu) substitutions, where the difference in molecular topology is more pronounced than with the alanine and butyrate system. While solid solutions consisting of L/L′ or DL/D′L′ leucine/methionine are unknown in the literature, successful use of this amino acid pair in quasiracemic10c and protein point mutation22 studies offers support to supramolecular methods seeking to exploit the structural commonality of the Met/Leu group. As shown in Fig. 3a, the previously reported structure of the L-leucine/D-methionine quasiracemate displays ordered components with near inversion symmetry. Can the structural features of this desymmetrized alignment tolerate chemical modification of the methionine and leucine components and still form co-crystals that adopt near inversion symmetry? In the case of our previously reported design strategy, such modifications include ‘fish-hook’ shaped frameworks that incorporate reactive sites and complementary non-covalent interactions to ensure the formation of robust motifs.17a
 | ||
| Fig. 3 Crystal structures of quasiracemates (A) L-leucine/D-methionine and (B) L-3/D-4 and (C) racemic compounds of 3 and 4. (atomic coordinates taken from ref. 10c). | ||
This investigation demonstrates how the topological similarity of methionine and leucine can be used to advantage to create reduced local and crystal symmetry. When applied to photoactive sulfonamidecinnamic acid frameworks the result is enantiocontrolled SCSC photodimerization reactions. Comparison of the crystal structures and solid-state reactivity of quasiracemic L-3/D-4 to its racemic and homochiral counterparts provides an important glimpse to the fundamental structural principles governing chiral transmission of desymmetrized assemblies to molecular crystal photochemical transformations.
Conformational tendencies of sulfonamidecinnamic acids
Fig. 3b and c show the anticipated ‘fish hook’ shaped molecules of 3 and 4 constructed from sulfonamidecinnamic acid decorated with Met and Leu amino acids organized in hydrogen-bonded dimers (ESI†). Though the strong penchant to form this conformation and ultimate dimer association is evident from similar sulfonamides previously reported and those described here, the observed outcome is somewhat unexpected given the conformational flexibility of the molecular framework. Because molecular shape plays a key role in the molecular recognition process of dimer formation, we then set out to compare the structural trends for the quasiracemate, racemates of 3 and 4, and L-3 to those in the extant database. Inspection of the CSD (Version 5.34) for arylsulfonamide crystal structures offered important insight to the structural trends of these sulfonamide systems. Fig. 4a shows this CSD search uncovered 2181 arylsulfonamides with a mean C–S–N–C (τ1) torsion angle of 67.4°. In a related search, the 70 carboxy decorated arylsulfonamides (X = CO2H) exhibit a mean S–N–C–CO2H (τ2) torsion value of 86.3° for the structures that cluster below 115° (Fig. 4B). Both mean torsion angles from these CSD searches (τ1 = 67.4° and τ2 = 86.3°) are comparable to those observed for the group of structures selected for this study – i.e., L-3/D-4, rac-3, rac-4, and L-3. The general leaning toward these conformational features coupled with two highly directional carboxyl–carboxyl nonbonded interactions provides a plausible argument for the driving force of dimer motif formation. | ||
| Fig. 4 Conformational comparison of τ1 and τ2 angles for (a) arylsulfonamides retrieved from the CSD database and (b) the crystal structures of 3–5 components used for this study (* denotes mean values). | ||
Interested in identifying the structural features that contribute to the desired molecular shape and dimer motif, we then turned our attention to preparing glycine derivative 5 where R = H. The two distinct phases of this compound (5-I and II) were accessed by recrystallization from CH2Cl2–MeOH and acetone–MeOH–H2O solutions. Both crystal structures display pronounced unfolding of the molecules of 5 with τ2 values approaching 180° (Fig 4D). Because the result is ‘L’ shaped component topologies, it is not surprising the desired supramolecular dimers give way to other motifs such as molecular chains. The 12 entry subset from the CSD search shows the impact of R/R′ = H to τ2 (Fig. 4B). Though this group of structures span a wide range of values (56–179°), the cluster at τ2 = 165–180° follows a similar conformational pattern seen in the structures of 5-I and II. In light of the quasiracemic (L-3/D-4), racemic (3 and 4), and achiral (5-I and -II) compounds included with the current study, the observed trends for τ1 and τ2 provide needed insight to understand the structural features responsible for the desired molecular topology and ultimate successful alignment of these sulfonamidecinnamic acids into photoreactive assemblies.
To date, we have reported a homologous family of structures constructed of components with R = methyl, ethyl, i-propyl, benzyl and for the current study hydrogen, isobutyl, and methylthioethyl.17 For each racemic and quasiracemic structure the molecules assemble in the crystal to give the expected ‘fish hook’ conformers. The structural deviations from this trend arise from use of frameworks that incorporate α-methylcinnamic acid and, as shown with this study the use of glycine (R = H). Close inspection of the structural features of these outliers offers critical insight to identify the structural boundaries of the preferred molecular shape. While the topology of the alpha substituted cinnamate likely originates from the steric bulk of the pendant methyl group, examples with glycine draw attention to the steering ability of the attached amino acid R group to sulfonamidecinnamic acid conformations. Rather than the opened framework observed for glycine 5-I and II, instances with R = alkyl consistently result in conformations where the intramolecularly aligned amino acid carboxyl and cinnamyl groups form nearly coplanar stacked molecular arrangements. Interestingly, the requisite fish hook conformer arises despite the use of a diverse set of substituents that vary from methyl to benzyl. As such, these studies seem to suggest R ≠ H as a critical factor for directing the shapes of these aryl sulfonamide frameworks.
UV induced crystal transformations
The preferred molecular conformations observed in the structures of quasiracemate L-3/D-4 and racemates 3 and 4 are well suited to explore solid-state photodimerization behavior. Each of these systems organize with the components forming supramolecular dimers due to complementary features of molecular shape and hydrogen-bond groups (Fig. 3B and C). In the case of quasiracemate L-3/D-4 the cinnamyl olefin groups are displaced by 3.92/3.97 Å. Similarly, compounds rac-3 and rac-4 align with favorable CC⋯CC interactions at 4.09 and 3.71 Å, respectively. Because such structures present model systems with minimal motion for investigating topochemically controlled [2 + 2] photoreactions,23 we then pursued these reactions using the established tail-irradiation technique to increase the opportunity for single-crystal-to-single-crystal (SCSC) transformations.24 In each case, illumination using a 200 W Xe(Hg) arc lamp equipped with a 360 nm cut-on optical filter resulted in the desired SCSC reactions. Fig. 5 shows the result of photoirradiating a crystal of L-3/D-4. During the course of the transformation, reaction progress was evaluated periodically by tracking the occupancy factors of the reactant and photodimer phases. After 22 hours of UV irradiation, the initial hydrogen-bonded dimer converted quantitatively to the [2 + 2] photoproduct with minimal decomposition of the crystalline sample.
 | ||
| Fig. 5 Single-crystal-to-single-crystal photodimerization of L-3/D-4 indicating reactant:photoproduct ratios (dashed lines indicate crystal structure minor component). | ||
The solid-state photocycloaddition of quasiracemate L-3/D-4 offers an interesting glimpse into the design and implementation of desymmetrized solid-state transformations. Molecular components for this system were engineered to incorporate the key elements of chirality, molecular topology, and reactive centers. When recrystallized, the blend of these structural features gives access to robust crystalline architectures capable of unique chemical outcomes. The infusion of chirality into this system starts with the handedness of components L-3 and D-4 that when assembled in the crystal form asymmetric supramolecular dimers. Owing to the predetermined alignment of these motifs, molecular and motif chirality then effectively translates to enantiocontrolled reaction products. By manipulating chiral transmission using designer synthons, such strategy offers considerable advantage for generating chiral assemblies capable of asymmetric transformations that could also benefit other material functions that require asymmetric molecular associations.
Extending the current investigation to include racemic and single-component homochiral systems provided a practical strategy to understand the structural trends and reactivity behavior of the sulfonamidecinnamic acid framework. As shown in Fig. 6, racemic phases of 3 and 4 both undergo photodimerization processes with similar outcomes to the UV reaction of L-3/D-4. However, because their dimeric assemblies are rigorously centrosymmetric, any UV initiated reactions that originate from these architectures lack a homochiral component to the photodimerization process. Our study of the photodimerization reaction of rac-4 indicated near convergence at 64% with retained crystallinity of the sample. By contrast crystals of leucine rac-3 fractured considerably during the course of the irradiation process. Inspection of Fig. 7 shows the overlay of the sulfonamide fragments present in the reactant and photoproduct phases. In the case of samples L-3/D-4 and rac-4 the conformational differences are minimal. However, the quality of X-ray data corresponding to irradiated rac-3 is greatly diminished due to considerable crystal degradation. One notable difference of rac-3 to the structures of L-3/D-4 and rac-4, and possible explanation for the decay in crystal quality of L-3, is the extensive movement of the leucine side-chain that accompanies the reaction process. Though such large displacements may be tolerable in some SCSC instances if molecular movement follows a concerted path, arguments of molecular motion have also been invoked to explain crystal degradation.24,25 As can be seen in the photographs depicted in Fig. 6, it seems that in the case of rac-3 the consequence of this motion gives rise to extensive crystal decomposition.
 | ||
| Fig. 6 UV irradiated rac-3 and rac-4 showing crystal structures of reactant (dashes) and photoproduct phases and samples before and after UV exposure. | ||
 | ||
| Fig. 7 Overlay of the L-3/D-4, rac-3, and rac-4 crystal structures showing the conformational preferences of the reactant (green) and photodimer (grey) phases. | ||
Examining homochiral L-3 provides an additional view of the utility of the sulfonamidecinnamic acid approach for generating chiral reactive supramolecular assemblies. The single-component system of L-3 crystallizes in space group P21 and favors the formation of catemeric hydrogen-bonded assemblies rather than the desired supramolecular dimers (Fig. 8). Despite the observed deviation from hydrogen-bonded pairs, the conformational parameters τ1 and τ2 for L-3 and other systems reported in Fig. 4 show a high degree of similarity. This supports the notion that the anticipated ‘fish hook’ molecular shapes are not solely controlled by motif selection, but rather likely derived from an assortment of factors that include enthalpically preferred conformations. For L-3, this conformation results in undulating chains where the two-fold screw related molecules link via carboxyl⋯carboxyl interactions. This motif extends along the b-axis with neighboring olefin groups skewed by 23.4° at distances of 4.04 and 4.36 Å. Though such an arrangement is less than ideal for photochemical studies, photoirradiated samples of L-3 indicated the formation of cyclobutane product. Initially, single crystals were processed as SCSC transformations. Reaction conversions could only be assessed to the ~20% conversion level via a SCSC reaction owing to sample degradation. Even so, the diffraction pattern corresponding to this sample clearly showed diffraction peaks consistent with a cyclobutane group forming between the olefin groups (Fig. 8, bottom). Processing a powdered sample of L-3 with unfiltered UV radiation provided further support for this transformation. The progress of the reaction was followed by 1H NMR and showed the appearance of signals at 4.04 and 4.51 ppm consistent with the cyclobutane Csp3H groups of the head-to-tail photoproduct (ESI, Fig. S1†).
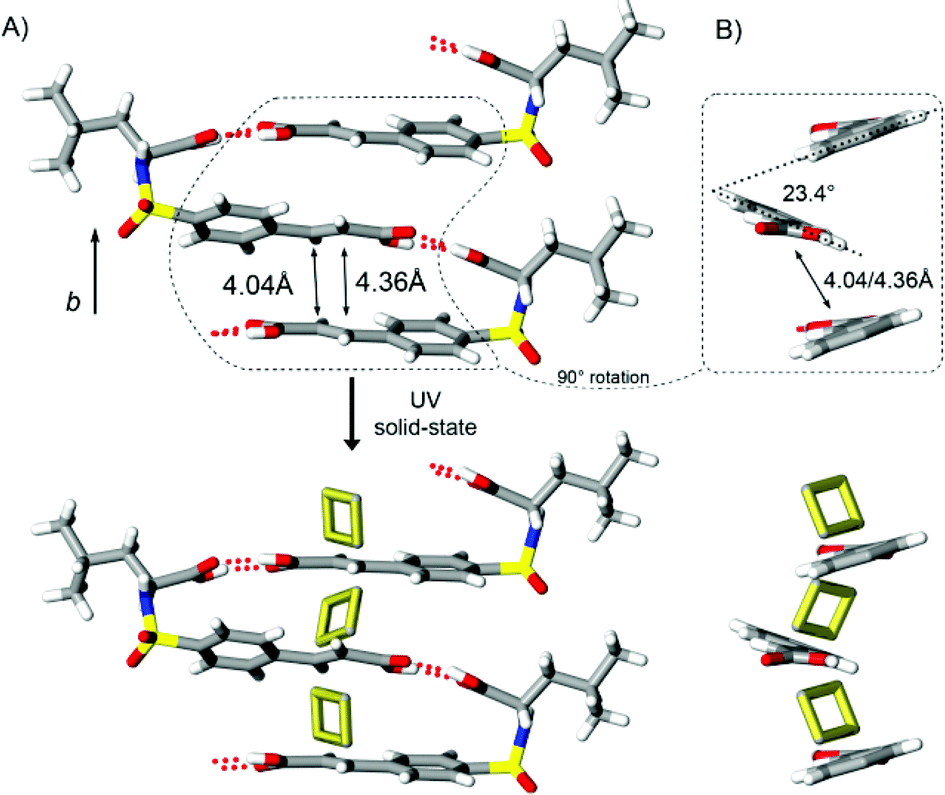 | ||
| Fig. 8 Crystal structures depicting the SCSC reaction of L-3 showing (A) catemeric hydrogen-bonded assemblies, olefin geometries, and photodimerization outcome and (B) skewed alignment of isolated cinnamoyl and cyclobutane groups. | ||
Conclusions
While photodimerization reactions in molecular crystals have received extensive attention in recent years, there remains a wealth of opportunities to explore untapped or lightly travelled areas of technological importance. The value of this familiarity is significant and can offer fertile ground for moving beyond conventional views and practices to develop research projects targeting current chemical challenges. In the case of UV induced solid-state [2 + 2] cycloadditions, much attention following Schmidt and Cohen's era23 has been directed to managing the alignment of reactive centers.19 This collective effort has helped pave the way for studies seeking to understand the complexity of reaction details26 and exploit the functionality of photodimerization products and processes.27 In the context of this contribution, imprinting homochirality onto these reactions has also emerged as an area of interest.17a,20,21 While this requires consideration of best practices for balancing supramolecular control, desymmetrized motifs, and chemical reactivity, strategies that successfully navigate such challenges will offer much needed entry and impetus to an area that holds importance to the scientific community.This work provides an effective approach for generating chiral photoreactive crystalline materials by exploiting the deliberate use of molecular topology. Building on the near inversion symmetry packing tendencies of amino acid quasiracemates, we derivatized the L-leucine and D-methionine pair with sulfonamidecinnamic acid groups resulting in ‘fish hook’ conformations consistent with similar structures in the extant database. As evident from the two crystal phases of glycine 5, the pendant R groups are necessary to achieve the desired molecular topology. This engineered shape coupled with two directional carboxyl⋯carboxyl interactions offers a strong driving force for the formation of reactive supramolecular dimers. In the case of quasiracemate L-3/D-4, these assemblies are rigorously noncentrosymmetric where the chirality of the components transfers to the cyclobutyl photoproduct processed as a UV initiated SCSC transformation. Similar crystal packing motifs and reaction outcomes were achieved for the racemic forms of 3 and 4 demonstrating the importance of best-fit scenarios directed by inversion related motifs. The homochiral single-component L-3 crystallized in space group P21 with catemeric motifs constructed from carboxyl⋯carboxyl hydrogen bonds. Despite the lack of supramolecular dimer formation, the observed motif catalyzes the reaction to give the head-to-tail photodimerization product.
Acknowledgements
This work was supported by the National Science Foundation (CHE-0957391 and CHE-0722547) and a Council on Faculty Research Grant from Eastern Illinois University (EIU). K. A. W. thanks the Cambridge Crystallographic Data Centre, Dr S. Henderson (host), and EIU for generous sabbatical support. We thank J. L. Horner for help with preliminary synthetic studies.Notes and references
- (a) H. X. Zhu, T. X. Fan and D. Zhang, On the Optimal Mechanical Properties of Hierarchical Biomaterials, in Hierarchically Structured Porous Materials: From Nanoscience to Catalysis, Separation, Optics, Energy, and Life Science, ed. B.-L. Su, C. Sanchez and X.-Y. Yang, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011 Search PubMed; (b) B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179 CrossRef CAS; (c) O. Parisa, I. Burgerta and P. Fratzla, MRS Bull., 2010, 35, 219 CrossRef; (d) E. Munch, M. E. Launey, D. H. Alsem, E. Saiz, A. P. Tomsia and R. O. Ritchie, Science, 2008, 322, 1516 CrossRef CAS PubMed; (e) R. Pfeifer, M. Lungarella and F. Iida, Science, 2007, 318, 1088 CrossRef CAS PubMed.
- (a) R. Hyde, P. M. Taylor and H. S. Hundal, Biochem. J., 2003, 373, 1 CrossRef CAS; (b) H. S. Hundal and P. M. Taylor, Am. J. Physiol.: Endocrinol. Metab., 2009, 296, E603 CrossRef CAS PubMed.
- P. E. Carrigan, P. Ballar and S. Tuzmen, Methods Mol. Biol., 2011, 700, 107 CAS.
- (a) S. Kawashima and M. Kanehisa, Nucleic Acids Res., 2000, 28, 374 CrossRef CAS PubMed; (b) K. Tomii and M. Kanehisa, Protein Eng., 1996, 9, 27 CrossRef CAS PubMed.
- (a) Y. Weisinger-Lewin, F. Frolow, R. K. McMullan, T. F. Koetzle, M. Lahav and L. Leiserowitz, J. Am. Chem. Soc., 1989, 111, 1035 CrossRef CAS; (b) J. L. Wang, Z. Berkovitch-Yellin and L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Sci., 1985, 41, 341 CrossRef; (c) L. Addadi, Z. Berkovitch-Yellin, N. Domb, E. Gati, M. Lahav and L. Leiserowitz, Nature, 1982, 296, 21 CrossRef CAS.
- P. Swaminathan and R. Srinivasan, J. Cryst. Mol. Struct., 1975, 5, 101 CrossRef CAS.
- B. Dalhus and C. H. Gorbitz, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 720 CAS.
- L. Addadi, A. Berkovitch-Yellin, I. Weissbuch, M. Lahav, L. Leiserowitz and S. Weinstein, J. Am. Chem. Soc., 1982, 104, 2075 CrossRef CAS.
- (a) Q. Zhang and D. P. Curran, Chem. – Eur. J., 2005, 11, 4866 CrossRef CAS PubMed; (b) A. G. Mitchell, J. Pharm. Pharm. Sci., 1998, 1, 8 CAS; (c) J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates and Resolutions, John Wiley and Sons, New York, 1981 Search PubMed; (d) E. L. Eliel and S. H. Wilen, Stereochemistry of Organic Compounds, Wiley, New York, 1994, p. 1267 Search PubMed.
- (a) C. H. Gorbitz, K. Rissanen, A. Valkonen and A. Husabo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, o267 Search PubMed; (b) B. Dalhus and C. H. Gorbitz, Acta Crystallogr., Sect. B: Struct. Sci., 1999, 55, 424 CrossRef PubMed; (c) B. Dalhus and C. H. Gorbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1547 Search PubMed; (d) C. H. Gorbitz, B. Dalhus and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8466 RSC; (e) B. Dalhus and C. H. Gorbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1105 Search PubMed; (f) G. S. Prasad and M. Vijayan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1991, 47, 2603 CrossRef.
- (a) M. Hendi, P. Hooter, V. Lynch, R. E. Davis and K. A. Wheeler, Cryst. Growth Des., 2004, 4, 95 CrossRef CAS; (b) B. Dalhus and C. H. Gorbitz, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 715 CAS.
- (a) D. E. Mortenson, K. A. Satyshur, I. A. Guzei, K. T. Forest and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 2473 CrossRef CAS PubMed; (b) K. Mandal, B. L. Pentelute, D. Bang, Z. P. Gates, V. Y. Torbeev and S. B. H. Kent, Angew. Chem., Int. Ed., 2012, 51, 6 Search PubMed.
- (a) G. Lautrette, B. Kauffmann, Y. Ferrand, C. Aube, N. Chandramouli, D. Dubreuil and I. Huc, Angew. Chem., Int. Ed., 2013, 52, 11517 CrossRef CAS PubMed; (b) C. Ebner, C. A. Muller, C. Markert and A. Pfaltz, J. Am. Chem. Soc., 2011, 133, 4710 CrossRef CAS PubMed; (c) F. Yang, J. J. Newsome and D. P. Curran, J. Am. Chem. Soc., 2006, 128, 14200 CrossRef CAS PubMed.
- (a) E. Palovics, J. Schindler, F. Faigl and E. Fogassy, Comprehensive Chirality, ed. E. M. Carreira and H. Yamamoto, Elsevier, Amsterdam, 2012, 8, 91–95 Search PubMed; (b) N. A. Shaye, S. Chavda, E. Coulbeck, J. Eames and Y. Yohannes, Tetrahedron: Asymmetry, 2011, 22, 439 CrossRef CAS PubMed; (c) F. Cardona, D. Lalli, C. Faggi, A. Goti and A. Brandi, J. Org. Chem., 2008, 73, 1999 CrossRef CAS PubMed.
- (a) S. Akine, S. Hotate, T. Matsumoto and T. Nabeshima, Chem. Commun., 2011, 47, 2925 RSC; (b) Q. Zhang, A. Rivkin and D. P. Curran, J. Am. Chem. Soc., 2002, 124, 5774 CrossRef CAS PubMed.
- (a) T. Jacobs, M. W. Bredenkamp, P. H. Neethling, E. G. Rohwer and L. J. Barbour, Chem. Commun., 2010, 46, 8341 RSC; (b) J. G. Nery, G. Bolbach, I. Weissbuch and M. Lahav, Chem. – Eur. J., 2005, 11, 3039 CrossRef CAS PubMed.
- (a) R. C. Grove, S. H. Malehorn, M. E. Breen and K. A. Wheeler, Chem. Commun., 2010, 46, 7322 RSC; (b) A. M. Lineberry, E. T. Benjamin, R. E. Davis, W. S. Kassel and K. A. Wheeler, Cryst. Growth Des., 2008, 8, 612 CrossRef CAS; (c) K. A. Wheeler, R. C. Grove, R. E. Davis and W. S. Kassel, Angew. Chem., Int. Ed., 2008, 47, 78 CrossRef CAS PubMed.
- (a) I. Halasz, Cryst. Growth Des., 2010, 7, 2817 CrossRef; (b) V. Enkelmann, G. Wegner, K. Novak and K. K. B. J. Wagener, J. Am. Chem. Soc., 1993, 115, 10390 CrossRef CAS.
- (a) K. Biradha and R. Santra, Chem. Soc. Rev., 2013, 42, 950 RSC; (b) G. K. Kole and J. J. Vittal, Chem. Soc. Rev., 2013, 42, 1755 RSC.
- (a) K. Tanaka, F. Toda, E. Mochizuki, N. Yasui, Y. Kai, I. Miyahara and K. Hirotsu, Angew. Chem., Int. Ed., 1999, 38, 3523 CrossRef CAS; (b) K. Tanaka, Molecules, 2012, 17, 1408 CrossRef CAS PubMed; (c) C. Yang, C. Ke, W. Liang, G. Fukuhara, T. Mori, Y. Liu and Y. Inoue, J. Am. Chem. Soc., 2011, 133, 13786 CrossRef CAS PubMed.
- (a) I. Weissbuch and M. Lahav, Chem. Rev., 2011, 111, 3236 CrossRef CAS PubMed; (b) I. Weissbuch, L. Leiserowitz and M. Lahav, Isr. J. Chem., 2011, 51, 1017 CrossRef CAS.
- (a) R. O. Cohen, G. Shen, J. H. Golbeck, W. Xu, P. R. Chitnis, A. I. Valieva, A. Van der Est, Y. Pushkar and D. Stehlik, Biochemistry, 2004, 43, 4741 CrossRef CAS PubMed; (b) L. A. Lipscomb, N. C. Gassner, S. D. Snow, A. M. Eldridge, W. A. Baase, D. L. Drew and B. W. Matthews, Protein Sci., 1998, 7, 765 CrossRef CAS PubMed; (c) D.-H. Kim, G. Edwalds-Gilbert, C. Ren and R.-J. Lin, Genetics, 1999, 153, 1105 CAS.
- (a) M. D. Cohen and G. M. J. Schmidt, J. Chem. Soc., 1964, 2000 RSC; (b) G. M. J. Schmidt, Pure Appl. Chem., 1971, 27, 647 CrossRef CAS.
- (a) J. B. Benedict and P. Coppens, J. Phys. Chem. A, 2009, 113, 3116 CrossRef CAS PubMed; (b) M. Kahn, V. Enkelmann and G. Brunklaus, Cryst. Growth Des., 2009, 9, 2354 CrossRef; (c) I. Abdelmoty, V. Buchholz, L. Di, V. Enkelmann, G. Wegner and B. M. Foxman, Cryst. Growth Des., 2005, 5, 2210 CrossRef CAS.
- G. Kaupp, Top. Curr. Chem., 2005, 254, 95 CAS.
- (a) T. V. Sreevidya, D.-K. Cao, T. Lavy, M. Botoshansky and M. Kaftory, Cryst. Growth Des., 2013, 13, 936 CrossRef CAS; (b) D.-K. Cao, T. V. Sreevidya, M. K. Botoshansky, G. Golden, J. B. Benedict and M. Kaftory, J. Phys. Chem. A, 2010, 114, 7377 CrossRef CAS PubMed; (c) J. B. Benedict and P. Coppens, J. Phys. Chem. A, 2009, 113, 3116 CrossRef CAS PubMed; (d) I. Turowska-Tyrk, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 670 Search PubMed.
- (a) S. Ghorai, J. C. Sumrak, K. M. Hutchins, D.-K. Bucar, A. V. Tivanski and L. R. MacGillivray, Chem. Sci., 2013, 11, 4304 RSC; (b) E. Elacqua, T. Friscic and L. R. MacGillivray, Isr. J. Chem., 2012, 52, 53 CrossRef CAS; (c) J. Stojakovic, B. S. Farris and L. R. MacGillivray, Chem. Commun., 2012, 48, 7958 RSC; (d) T. D. Hamilton, D.-K. Bucar, J. Baltrusaitis, D. R. Flanagan, Y. Li, S. Ghorai, A. V. Tivanski and L. R. MacGillivray, J. Am. Chem. Soc., 2011, 133, 3365 CrossRef CAS PubMed; (e) E. Elacqua and L. R. MacGillivray, Eur. J. Org. Chem., 2010, 36, 6883 CrossRef; (f) J. W. Chung, Y. You, H. S. Huh, B.-K. An, S.-J. Yoon, S. H. Kim, S. W. Lee and S. Y. Park, J. Am. Chem. Soc., 2009, 131, 8163 CrossRef CAS PubMed; (g) P. Zhao, C. F. Fang, C. J. Xia, Y. M. Wang, D. S. Liu and S. J. Xie, Appl. Phys. Lett., 2008, 93, 013113 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, photochemical studies, and full crystal structure details and tables for L-3/D-4, rac-3, L-3, rac-4, 5-I and 5-II. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra47270a |
| This journal is © The Royal Society of Chemistry 2014 |