DOI:
10.1039/C3RA47234E
(Paper)
RSC Adv., 2014,
4, 9261-9270
Selective and sensitive electrochemical detection of dopamine based on water-soluble porphyrin functionalized graphene nanocomposites
Received
3rd December 2013
, Accepted 22nd January 2014
First published on 22nd January 2014
Abstract
A biosensor, based on a water-soluble porphyrin-reduced graphene oxide (RGO) nanocomposite synthesized by simultaneous covalent and non-covalent strategies through aromatic π–π stacking and the formation of chemical bonds, was prepared for selective and sensitive detection of dopamine (DA). Compared with graphene or porphyrin alone, porphyrin-RGO nanocomposites exhibited unique advantages for the detection of DA in the presence of interfering substances such as uric acid (UA) and ascorbic acid (AA). The cyclic voltammetry (CV) curves indicated that the porphyrin-RGO modified glassy carbon electrode (GCE) had larger active area and better electrochemical catalytic activity which could attribute to the π–π stacking and the electrostatic attraction between positive charged DA and negative charged porphyrin-RGO, which can accelerate the electron transfer and weaken the oxidation of AA/UA on the porphyrin-RGO/GCE. Differential pulse voltammetry (DPV) was used for the quantitative detection of DA. The peak currents increased linearly with the increasing concentration of DA in the range of 1 × 10−6 to 7 × 10−5 M, and the limit of detection (LOD) (S/N = 3) was estimated to be 9.45 × 10−9 M. More importantly, the biosensor exhibited good stability and reproducibility, and would provide a superior platform in the biological analysis.
1. Introduction
Dopamine (DA), as an important catecholamine neurotransmitter, plays a vital role in mammalian central nervous, cardiovascular, renal and hormonal systems.1,2 The abnormal levels of DA can cause neurological disorders, such as Parkinson's disease and Schizophrenia.3 Therefore, it is very necessary to detect precisely the concentration of DA in the biological systems. At present, electrochemical technologies were paid more considerable attention for the detection of DA due to the advantages of quickness, portability, high-sensitivity and low-cost. Based on the electroactive nature of DA, voltammetry4–6 and chronoamperometry7 were used frequently to detect DA in the past two decades. However, ascorbic acid (AA) and uric acid (UA) were the main interfering substances during the detection of DA because of co-existence with DA in the real biological samples and the similar oxidation peak potential resulting in the overlap of peaks. To overcome this problem and develop selective and sensitive methods for the detection of DA, many electroactive materials, possessing catalytic effects on oxidation of DA, were employed to obtain the modified electrodes.
Graphene, a two-dimensional (2D) one atom thick nanomaterial consisting of sp2-hybridized carbon, was used as an ideal modification material in chem-/bio-sensors widely8,9 owing to its unique properties, such as high surface area,10 excellent electrical conductivity,11 and well electrocatalytic activity.12 Currently, many electrochemical sensors,13,14 using graphene, especially functionalized graphene as the sensing materials, were developed for molecule detection. For instance, graphene oxide,15 reduced graphene oxide (RGO),16 graphene/metal nanoparticle,17,18 graphene/metal oxide19 and graphene/metal oxide/metal nanoparticle20 were applied to selectively detect DA, showing the advantages of low-cost, high-sensitivity and selectivity. Such as, a biosensor, based on dodecyl sulfonate-graphene nanosheet/SnO2 hybrid nanocomposites (SDS-GN/SnO2), was developed for the selective detection of DA.19 It exhibited well reproducibility and sensitivity, and the LOD was as low as 8.0 × 10−8 M. Using the nanocomposite of ferrocene thiolate stabilized Fe3O4/Au nanoparticles with graphene sheet as the sensing material, a double signal amplification for ultrasensitive and selective detection of DA was fabricated with the LOD of 1.0 × 10−7 M.20
Porphyrin, an anion compound with catalytic effect on the oxidation of positive charged DA, has been used to prepare modified electrodes for the selective detection of DA. Wu et al. presented an electrochemical sensor based on meso-tetra(4-carboxyphenyl) porphine-functionalized graphene (TCPP/CCG), synthesized by non-covalent strategy. It can be applied to the selective determination of DA and the LOD was 1.0 × 10−8 M.21 However, during the non-covalent functionalization, it was found that there were poor electrochemical responses for DA, non-uniform dispersions and low loading amounts of porphyrin on the RGO. Here, we use protoporphyrin IX to modify RGO by simultaneous covalent and non-covalent strategies through aromatic π–π stacking and formation of amide bonds between porphyrin and graphene (as shown in Scheme 1). The as-obtained porphyrin-RGO nanocomposites possessed better water-soluble, electrochemical activities, uniform dispersions and high loading amounts of porphyrin on the RGO. Compared with bare GCE, single porphyrin or graphene modified electrodes, porphyrin-RGO/GCE had larger reaction surface area and better CV and DPV responses for DA in the present of interfering substances UA and AA. This electrochemical platform of porphyrin-RGO/GCE, exhibited well selectivity and sensitivity for the quantitative detection of DA, with the LOD 9.45 × 10−9 M.
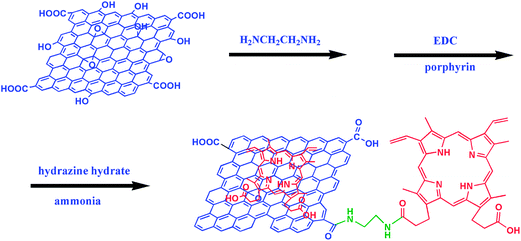 |
| | Scheme 1 The synthesis procedure of porphyrin-RGO nanocomposite. | |
2. Experimental
2.1. Reagents
All reagents used in this work were of analytical grade and employed without further purification. Nature graphite powders used for preparing GO were available from Duratight Sealing Product Co., Ltd. Qingdao, China. Protoporphyrin IX (95%) was purchased from Sigma-Aldrich. DA was purchased from Aladdin Chemistry Co., Ltd. AA was obtained from Sinopharm Chemical Reagent Co., Ltd. UA was purchased from J&K scientific Co., Ltd. DA, AA and UA solutions were prepared fresh prior to use. 0.1 M phosphate buffer solution (PBS) was prepared by mixing appropriate amounts of Na2HPO4 and NaH2PO4. Unless otherwise stated, all solutions were prepared with double-distilled water.
2.2. Preparation of porphyrin-RGO
GO prepared by a modified Hummers' method according to our early report22 (40 mg) and ethylenediamine (10 mL) were dispersed in deionized water (30 mL) by mild sonication for 30 min. The reaction flask was stirred for 10 h at 90 °C. Then, 20 mL of 1 mg mL−1 porphyrin solution containing 100 μL of 1 M NaOH, and EDC (6 mg) were added in the above solution at 90 °C for another 10 h. Finally, hydrazine hydrate (40 μL) and ammonia (800 μL) were added to the reaction flask at 95 °C for 3 h to reduce GO. After the reaction, the mixture was centrifuged at 8000 rpm for 15 min and then washed by deionized water to remove the un-reacted porphyrin and finally dried under vacuum at 70 °C to yield porphyrin-RGO.
2.3. Preparation of the modified GCE
Porphyrin-RGO (1 mg) was dispersed in 1 mL double-distilled water under ultrasonic agitation. Then, 10 μL of 1 M NaOH was added into the above solution and sonicated for 30 min to get a homogeneous solution with a concentration of approximately 1 mg mL−1. Prior to the surface modification, the bare GCE (Φ = 3 mm) was polished with 0.05 μm alumina powder and rinsed thoroughly with doubly distilled water. Then, it was cleaned successively with anhydrous ethanol and doubly distilled water in an ultrasonic bath and dried under nitrogen. A porphyrin-RGO/GCE was obtained by coating 5 μL porphyrin-RGO homogeneous solution on an electrode and dried in air. For comparison, RGO/GCE and porphyrin/GCE were prepared in the same manner. Finally, the modified electrodes were activated by several successive scans with a scan rate of 300 mV s−1 in 0.1 M PBS (pH = 7) until a steady cyclic voltammogram.
2.4. Detection of dopamine
20 mL 0.1 M PBS containing different concentrations of DA was added into the electrochemical cell. CV was carried out at a scan rate of 50 mV s−1. DPV measurements were used for the quantitative determination of DA under the following parameters: potentials range from −0.4 to 0.6 V; pulse width: 200 ms; pulse height: 50 mV; step height: 5 mV.
2.5. Characterization
The microstructures of GO and porphyrin-RGO were observed by transmission electron microscopy (TEM, Tecnai F20). For imaging, the samples were dropped from suspension onto micro-grids and dried under an infrared lamp. FT-IR analysis were recorded on a Perkin-Elmer spectrometer using a disc of KBr. Raman spectroscopic analysis was carried out on a Lab RAM HR 800UV (HORIBA Jobin Yvon, France) with an excitation laser of 532 nm. Thermal gravimetric analysis (TGA) was carried out by using a thermogravimetric analyzer (Perkin Elmer) at a heating rate of 10 °C min−1 from 20 to 800 °C in nitrogen. X-ray photoelectron spectroscopy (XPS) was measured on a VG multilab 2000 (Thermo electron Corporation). UV-vis absorption was carried out with a shimadzu-UV2550 Spectrophotometer, and fluorescence measurements were performed with FP-6500, Jasco. All electrochemical measurements were carried out on an IM6 electrochemical workstation with a standard three-electrode system. A bare or modified GCE (Φ = 3 mm) served as a working electrode; a platinum electrode and a saturated calomel electrode (SCE) were used as the counter electrode and the reference electrode, respectively.
3. Results and discussion
3.1. Characterization of porphyrin-RGO
TEM measurement was done to observe the morphologies of GO and porphyrin-RGO. Fig. 1a showed that the as-obtained GO sheets were homogeneous and quite smooth. As shown in Fig. 1b, after porphyrin was attached to RGO, it was found the surfaces became wrinkled and folded with porphyrin particles which were dispersed uniformly on the sheets of RGO.
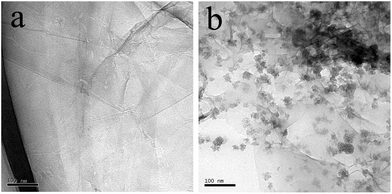 |
| | Fig. 1 TEM images of (a) GO and (b) porphyrin-RGO. | |
Fig. 2a showed the FTIR spectra of porphyrin, RGO and porphyrin-RGO. The peak of RGO at 1630 cm−1, which was associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of –COOH group, almost disappeared after the chemical reduction of GO. However, the intensity of the CO peak in porphyrin-RGO at 1630 cm−1 increased greatly due to the presence of CO in the –NHCO–, and the peak at 1260 cm−1 was attributed to the vibration of amide C–N. All results indicated that porphyrin was covalently functionalized on the surface of graphene. Raman spectrum is an effective tool to identify graphene-based materials. As shown in Fig. 2b, we compared the Raman spectra of GO, RGO and porphyrin-RGO. The Raman spectrum of GO showed two broad peaks at 1335 and 1590 cm−1, which corresponded to the D- and G-bands. The ratio of the intensities of D and G bands (ID/IG) is usually used to characterize the level of chemical functionalization in graphene-based materials. After chemical reduction, RGO also contained D- and G-bands, with an increased ID/IG (1.34) compared with GO (ID/IG = 0.95), which indicated that the average size of sp2 domains was decreased.23 For porphyrin-RGO, the ID/IG (1.57) was larger than GO and RGO, which was attributed to an increasing number of sp3 carbons formed on the graphene during the functionalization.13
O stretching vibration of –COOH group, almost disappeared after the chemical reduction of GO. However, the intensity of the CO peak in porphyrin-RGO at 1630 cm−1 increased greatly due to the presence of CO in the –NHCO–, and the peak at 1260 cm−1 was attributed to the vibration of amide C–N. All results indicated that porphyrin was covalently functionalized on the surface of graphene. Raman spectrum is an effective tool to identify graphene-based materials. As shown in Fig. 2b, we compared the Raman spectra of GO, RGO and porphyrin-RGO. The Raman spectrum of GO showed two broad peaks at 1335 and 1590 cm−1, which corresponded to the D- and G-bands. The ratio of the intensities of D and G bands (ID/IG) is usually used to characterize the level of chemical functionalization in graphene-based materials. After chemical reduction, RGO also contained D- and G-bands, with an increased ID/IG (1.34) compared with GO (ID/IG = 0.95), which indicated that the average size of sp2 domains was decreased.23 For porphyrin-RGO, the ID/IG (1.57) was larger than GO and RGO, which was attributed to an increasing number of sp3 carbons formed on the graphene during the functionalization.13
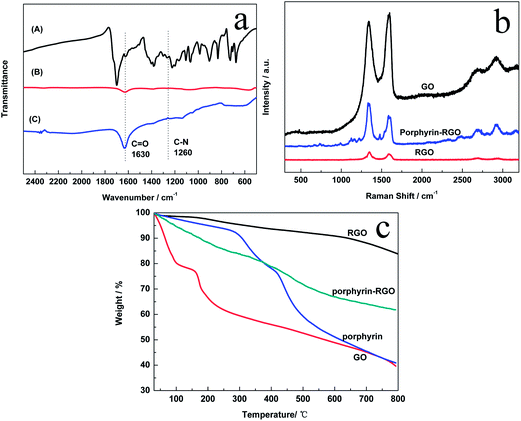 |
| | Fig. 2 (a) FTIR spectra of porphyrin (A), RGO (B) and porphyrin-RGO (C). (b) Raman spectra of GO, RGO and porphyrin-RGO. (c) TGA curves of GO, RGO, porphyrin and porphyrin-RGO. | |
Fig. 2c showed the TGA curves of RGO, porphyrin-RGO, porphyrin and GO. It could be seen that GO suffered from a 20% mass loss below 100 °C due to the removal of the absorbed water. The weight reduction between 100 to 200 °C was attributed to the pyrolysis of the oxygen-containing functional groups, and the carbon skeleton of GO started to decompose until 200 °C. Compared with GO, porphyrin was more thermally stable because of the existence of conjugated structure and the weight loss happened until 280 °C. The thermal stability of RGO was improved obviously with only 15% weight loss up to 800 °C due to the removal of oxygen-containing functional groups after chemical reduction. However, porphyrin-RGO only retained about 62% of the original mass compared with 85% of RGO when heated to 800 °C, which suggested that porphyrin molecules modified at the edges of the RGO sheets was about 23%. The successful functionalization of RGO was further verified.
To investigate the chemical composition and verify the functionalization of RGO further, X-ray photoelectron spectroscopic (XPS) measurements were carried out on GO and porphyrin-RGO. As can be seen from Fig. 3a, the survey spectra of GO showed the C1s and O1s peaks, with the binding energy of 284.5 eV and 531.9 eV, respectively. Compared with GO, the XPS spectra for porphyrin-RGO showed an obvious N1s peak at 401.6 eV and the nitrogen content was calculated to be about 6.3 at%. As shown in Fig. 3b, the C1s spectrum of porphyrin-RGO could be divided four obvious peaks, the main peak at 284.8 eV was attributed to CC bonds, the peaks at 285.8 eV and 287.5 eV corresponded to CN and C–N bonds,24 respectively, while the peak at 289.3 eV was attributed to O–CO. The high-resolution N1s spectrum was employed to confirm the binding configurations of N atoms in porphyrin-RGO. Fig. 3c displayed that, the main peak at 399.5 eV corresponded to pyrrole-like nitrogen, while the two small speaks at 398.0 eV and 401.1 eV corresponded to pyridine-like nitrogen.25 All the above analysis proved that porphyrin had been functionalized to RGO by covalent and non-covalent strategies.
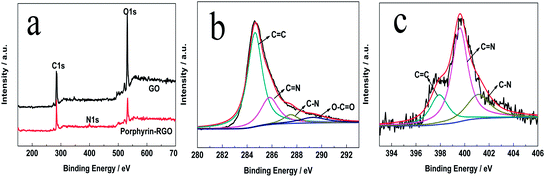 |
| | Fig. 3 (a) XPS survey spectra of GO and porphyrin-RGO, High-resolution C1s (b) and N1s (c) peaks of porphyrin-RGO. | |
Fig. 4a showed UV-vis absorption spectra of porphyrin, RGO and porphyrin-RGO. RGO showed a strong absorption peak at 285 nm, which corresponded to the n–π* transition of the CO bonds. The porphyrin spectrum contained a strong Soret absorption at 378 nm and weak Q-bands between 500 and 700 nm. For porphyrin-RGO, it exhibited a broad absorption at 283 nm, which should be the corresponding peak of RGO at 285 nm. Meanwhile, compared with porphyrin, a bathochromic shift of Soret band with 50 nm (from 378 to 428 nm) and almost disappeared Q-bands were observed, which were attributed to the strong π–π stacking interaction between RGO and porphyrin. Fig. 4b showed the UV-vis absorption spectra of solutions of porphyrin-RGO with different concentrations. Well linear relationships between the maximum absorption value (428 nm and 283 nm) and the concentrations were observed for the moieties of porphyrin and graphene in this hybrid, which obeyed Beer's law at low concentrations. It was used to determine the solubility of modified graphene. All those results indicated that porphyrin was functionalized on the graphene successfully and porphyrin-RGO could be dispersed homogeneously in water. Fig. 4c showed the dispersion ability of GO, RGO and porphyrin-RGO in water intuitively. While GO solution was yellow and RGO deposited at the bottom of the aqueous solution within 10 min, porphyrin-RGO exhibited a homogeneous dispersion in water for a long time, which proved again that porphyrin improved the dispersion and solubility of RGO in water. In addition, a strong fluorescence emission peak was observed in porphyrin solution under the excitation at 405 nm (Fig. 4d). As to porphyrin-RGO, the fluorescence was quenched absolutely with quenching efficiency of 100% due to the occurrence of electron transfer from porphyrin to RGO.
 |
| | Fig. 4 (a) UV-vis absorption of porphyrin, RGO and porphyrin-RGO. (b) UV-absorption spectra of porphyrin-RGO dispersed in double-distilled water with different concentrations. The plots of absorption of porphyrin and graphene moieties versus concentrations were inseted in (A) and (B). (c) The photographs of GO (A), RGO (B) and porphyrin-RGO (C) in water. (d) Fluorescence spectra of porphyrin and porphyrin-RGO. | |
3.2. Electrochemical behavior of porphyrin-RGO/GCE
The surface behavior of modified GCE was investigated by CV in the freshly prepared solution of 0.1 M KNO3 containing 5 mM Fe(CN)63−/4−. Fig. 5a displayed the CV responses of bare GCE, porphyrin, porphyrin-RGO and RGO modified GC electrodes. As compared with bare GCE (Ipa = 49 μA, ΔEp = 93 mV), a pair of relatively weak redox peak with a larger peak potential difference was observed on the porphyrin modified GCE (Ipa = 43 μA, ΔEp = 349 mV). However, after adding RGO, improved redox peak currents and smaller peak potential difference occurred on the RGO/GCE (Ipa = 73uA, ΔEp = 87 mV) and porphyrin-RGO/GCE (Ipa = 71 μA, ΔEp = 138 mV), which indicated that the incorporation of graphene increased the reaction surface area of electrode and reduced the electron transfer resistance,26,27 which could attribute to the better electrical conductivity of graphene, promoting the electron exchange between the electrochemical probe [Fe(CN)6]3−/4− and the electrode. Although the values of redox currents on porphyrin-RGO/GCE were smaller relative to RGO/GCE, resulting from the poor conductivity of DA, it was much larger than bare GCE. All above results revealed that the combination of porphyrin and RGO might provide necessary conduction bridge and obtain a good electrochemical catalytic behavior.
 |
| | Fig. 5 (a) CVs measured with bare GCE, porphyrin, porphyrin-RGO and RGO modified GCE in the solution of 0.1 M KNO3 containing 5 mM Fe(CN)63−/4−. (b) CVs for 100 μM DA on bare GCE, porphyrin/GCE, RGO/GCE and porphyrin-RGO/GCE in 0.1 M PBS (pH = 7.0) with a scan rate of 50 mV s−1, Inset: the comparison between bare GCE and porphyrin/GCE. | |
Fig. 5b showed the CV responses for 100 μM DA on the bare GCE, porphyrin/GCE, RGO/GCE and porphyrin-RGO/GCE. No obvious redox peaks were obtained on the bare GCE and a pair of weak redox peak occurred on the porphyrin/GCE with the Ipa of 1.53 μA and Ipc of 1.31 μA. However, a pair of stronger redox peaks was observed on RGO/GCE and porphyrin-RGO/GCE, respectively. Although the peak potential difference (as shown in Table 1) was almost the same, the intensity of peak current on porphyrin-RGO/GCE (14.1 μA) was two times higher than that of RGO/GCE (6.9 μA), suggesting a much larger active area.
Table 1 Electrochemical parameters of 100 μM DA on the different electrodes
| Electrodes |
Epa/V |
Epc/V |
ΔEp/V |
Ipa/μA |
Ipc/μA |
| Bare GCE |
0.254 |
0.076 |
0.178 |
0.75 |
0.28 |
| Porphyrin/GCE |
0.239 |
0.164 |
0.075 |
1.53 |
1.31 |
| RGO/GCE |
0.182 |
0.113 |
0.069 |
6.90 |
3.85 |
| Porphyrin-RGO/GCE |
0.194 |
0.124 |
0.070 |
14.13 |
9.10 |
Since DA often co-exist with AA/UA in biological systems, it is necessary to detect the DA selectively in the presence of AA and DA. Fig. 6a showed that DA, AA and UA had different electrochemical behaviors on the porphyrin-RGO/GCE. For example, a pair of well-defined and strong redox peaks was observed in the CV response of DA, which attributed to that the π–π stacking and the electrostatic attraction between positive charged DA and negative charged porphyrin-RGO, accelerating the electron transfer. However, no peak and a very weak oxidation peak with a separated potential, occured in the CVs of AA and UA, respectively, which attributed to that the weak π–π stacking interaction and the electrostatic repulsion between negative charged AA/UA and porphyrin-RGO.21 Fig. 6b illustrated the DPVs of DA, AA and UA, and the results were consistent with that of CVs. All these results proved that porphyrin-RGO could be applied to selectively detect DA.
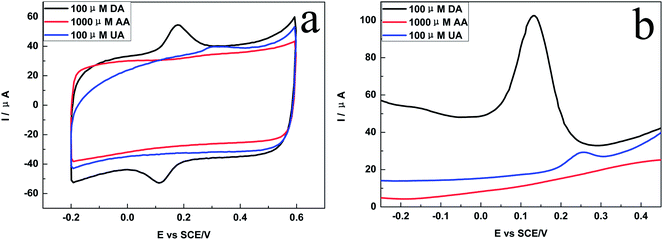 |
| | Fig. 6 (a) CVs and (b) DPVs of 100 μM DA, 1000 μM AA and 100 μM UA on the porphyrin-RGO/GCE in PBS (pH = 7.0). | |
The reaction kinetic of DA on the porphyrin-RGO/GCE was investigated by studying the effects of the scan rate on the redox peak currents (Ipa: anodic peak current and Ipc: cathodic peak current). Fig. 7a showed the CV responses of DA with the scan rates from 10 to 100 mV s−1 and Fig. 7b displayed intuitively that the current peak was proportional to the square root of the scan rate. This result suggested the electrochemical redox behavior of DA on the surface of porphyrin-RGO/GCE was a diffusion-controlled process in the low scan rate of 10–100 mV s−1, which predicted that DA could easily diffuse through the solution to the surface of porphyrin-RGO/GCE25 and there was sufficient time for the porphyrin-RGO film to behave as a three-dimensional electrode.21
 |
| | Fig. 7 (a) Different scan rates of 100 μM DA using porphyrin-RGO/GCE in the PBS. (b) The relation between the peak current versus the square root of the scan rate. | |
3.3. The effect of pH on the electrochemical behaviors
The electrochemical redox behaviors of DA at the porphyrin-RGO/GCE in the PBS with different pH values (from 5.00 to 9.00) were investigated by CV. As displayed in Fig. 8a, the pH of PBS had a significant influence on both the peak potentials and peak currents of DA. Fig. 8b showed that the peak currents reached a maximum at pH = 7.0, and it was selected as the optimum pH for the subsequent measurements to obtain a higher sensitivity. Meanwhile, the peak potentials shift negatively with the increase of pH from 5.00 to 9.00. A well linear relationship between the anodic peak potential (Epa) and pH was obtained with the equation Epa = 0.6036 − 0.0586pH (R2 = 0.990). The slope of −58.6 mV per pH was very close to the theoretical value (−59 mV per pH), which indicated that the identical numbers of protons and electrons were involved in the electrochemical redox process of DA. This was consistent with the former report.4
 |
| | Fig. 8 (a) The effect of pH on the CV responses of 100 μM DA in the PBS, (b) the relationship of peak potential (red) and peak current (blue) against pH. | |
3.4. Detection of DA
Porphyrin-RGO/GCE was applied for the quantitative detection of DA by DPV. Fig. 9a showed the DPV responses of different concentrations of DA on the porphyrin-RGO/GCE in the PBS (pH = 7.0). The results indicated that the peak currents enhanced with the addition amount of DA, and it predicted that the porphyrin-RGO/GCE could be applied to the quantitative detection of DA. Fig. 9b displayed a well linearity between peak currents and DA concentrations obtained in the range from 1 × 10−6 to 7 × 10−5 M. The linearization equation for DA was y = 1.0165x + 3.7608 (x: concentration/μM, y: current/μA), with the correlation coefficient of 0.998. The limit of detection (LOD) was calculated to be 9.45 × 10−9 M based on a signal-to-noise ratio of 3 (S/N = 3). It is worth to note that the LOD and sensitivity were well comparable and even better to the most of the reported methods (as shown in Table 2).
 |
| | Fig. 9 (a) DPV responses of DA for different concentrations (from bottom to top: 1 × 10−6, 2 × 10−6, 4 × 10−6, 6 × 10−6, 8 × 10−6, 1 × 10−5, 3 × 10−5, 5 × 10−5, 7 × 10−5 M) on the porphyrin-RGO/GCE in 0.1 M PBS (pH = 7.0). (b) The linear relationship between peak currents and the concentrations of DA. | |
Table 2 Comparison of different modified electrodes for the detection of DAa
| Electrode substrate |
Method |
Linear detection range (10−6 M) |
Limit of detection (10−6 M) |
Ref. |
| MIPs: molecularly imprinted polymers; SDS: sodium dodecyl sulfonate; Fc-SH: ferrocene thiol; HAu: hollow gold; CS: chitosan; PDDA: poly(diallyldimethylammonium chloride); TCPP: meso-tetra(4-carboxyphenyl) porphine; MWCNT: multiwall carbon nanotubes. |
| GO/GCE |
DPV |
1–15 |
0.27 |
15 |
| GO/SiO2-MIPs/GCE |
Chronoamperometry |
0.05–160 |
0.03 |
7 |
| Cu2O/RGO/GCE |
CV |
0.1–10 |
0.01 |
4 |
| SDS-RGO/SnO2/GCE |
DPV |
0.1–10 |
0.08 |
19 |
| Fe3O4@Au–S–Fc/RGO-CS/GCE |
DPV |
0.5–50 |
0.02 |
20 |
| HAu-RGO/GCE |
DPV |
0.08–600 |
0.05 |
17 |
| PdNPs/RGO/CS/GCE |
DPV |
0.5–15 |
0.1 |
6 |
| Pd3Pt1/PDDA-RGO/GCE |
DPV |
4–200 |
0.04 |
18 |
| TCPP/RGO/GCE |
DPV |
0.01–5 |
0.01 |
21 |
| Sulfonated-RGO/GCE |
DPV |
0.2–20 |
0.02 |
28 |
| RGO-MWNTs-PTA/GCE |
DPV |
0.5–20 |
1.14 |
29 |
| Porphyrin-RGO |
DPV |
1–70 |
0.0095 |
This work |
3.5. Interference study
As we know, AA and UA often co-exist with DA in the biological systems and their concentrations are usually much higher than that of DA. Therefore, it is very necessary to study the interference for the detection of DA. Fig. 10a showed the DPVs of different concentrations of DA on the porphyrin-RGO/GCE in the presence of 100 μM AA, the result presented no peak current of AA was observed in the potential ranges of −0.2 to 0.5 V. The linearization equation was y = 0.9868x + 6.2693 (x: concentration/μM, y: current/μA) with the correlation coefficient of 0.998 (Fig. 10b) and the LOD was evaluated to be 9.83 × 10−9 M based on a signal-to-noise ratio of 3, almost being consistent with the LOD (9.45 × 10−9 M) of single DA, which suggested that AA has no interference on the detection of DA. Fig. 10c illustrated the DPVs of different concentrations of DA in the presence of 100 μM UA and the peak currents of UA (E = 0.25 V) changed slightly during the redox of DA. The linearization equation was y = 0.8636x + 1.3078 (x: concentration/μM, y: current/μA) with the correlation coefficient of 0.994 (Fig. 10d) and the LOD was calculated to be 1.14 × 10−8 M based on S/N = 3, closing to that of single DA (9.45 × 10−9 M), which indicated that the interference of UA could be ignored. All these results showed that porphytin-RGO/GCE was well responsed for the determination of DA in the presence of interfering substances such as UA or AA.
 |
| | Fig. 10 DPV responses of DA for different concentrations (from bottom to top: 1 × 10−6, 2 × 10−6, 4 × 10−6, 6 × 10−6, 8 × 10−6, 1 × 10−5, 3 × 10−5, 5 × 10−5, 7 × 10−5 M) in the presence of AA of 100 μM (a) and the corresponding linear relationship between peak currents and the concentrations of DA (b). The DPV responses with UA of 100uM (c) and the corresponding relationship (d). | |
3.6. Evaluation of stability and reproducibility
A series of repetitive measurements (7 times) of DPV response for 70 μM DA in 0.1 M PBS were performed to further evaluate the stability of porphyrin-RGO/GCE (Inset of Fig. 11a). As shown in Fig. 11a, the highly reproducible DPV currents were observed with a relative standard deviation (RSD) of 0.96%. Meanwhile, we compared the responses of cycle 30 with the cycle 1 (Fig. 11b), the relative error was 2.38%. Therefore, the porphyrin-RGO nanocomposite modified electrode has an excellent stability for the repetitive DPV measurements. Meanwhile, seven different electrodes prepared were used to estimate the reproducibility, and the RSD of DPV current responses obtained was within 5%, indicating the acceptable reproducibility. In addition, we found that porphyrin-RGO/GCE could be repeatedly used for a long time without regenerating or reactivating the surface during the process of successive determinations.
 |
| | Fig. 11 (a) The stability of repetitive measurements (7 times) of DPV response for 70 μM DA on the porphyrin-RGO/GCE in 0.1 M PBS (pH = 7.0). Inset: the DPV responses of 70 μM DA under 7 times scans. (b) The DPV responses of cycle 1 and 30. | |
4. Conclusions
We synthesized porphyrin-RGO nanocomposites by simultaneous covalent and non-covalent strategies. Porphyrin-RGO possessed the unique advantages of porphyrin and graphene, and was used as the sensing material for the detection of DA. The results of electrochemical measurements suggested that porphyrin-RGO modified GCE had large active area and well electrochemical catalytic activity, and could be applied to the sensitive and selective determination of DA in the presence of interfering substances such as UA and AA. This result attributed to the synergistic effects from the excellent electrical conductivity of graphene and the catalytic effect on the oxidation of positive charged DA of porphyrin. The π–π stacking and the electrostatic attraction between positive charged DA and negative charged porphyrin-RGO could accelerate the electron transfer, and the electrostatic repulsion between negative charged AA/UA and porphyrin-RGO could weaken the oxidation of AA and DA on the porphyrin-RGO/GCE.
Acknowledgements
This work was supported by National Natural Science Foundation (51272071), Ministry of Education (20114208110005), Hubei Provincial Department of Education (D20111002, B2011802), and Wuhan Science and Technology Bureau (201271130447), China.
References
- R. K. Shervedani, S. M. Siadat-Barzoki and M. Bagherzadeh, Electroanalysis, 2010, 22, 969–977 CrossRef CAS.
- G. Z. Hu, D. P. Zhang, W. L. Wu and Z. S. Yang, Colloids Surf., B, 2008, 62, 199–205 CrossRef CAS PubMed.
- S. Sansuk, E. Bitziou, M. B. Joseph, J. A. Covington, M. G. Boutelle, P. R. Unwin and J. V. Macpherson, Anal. Chem., 2013, 85, 163–169 CrossRef CAS PubMed.
- F. Y. Zhang, Y. J. Li, Y. E. Gu, Z. H. Wang and C. M. Wang, Microchim. Acta, 2011, 173, 103–109 CrossRef CAS.
- S. M. Li, S. Y. Yang, Y. S. Wang, C. H. Lien, H. W. Tien, S. T. Hsiao, W. H. Liao, H. P. Tsai, C. L. Chang, C. C. Ma and C. C. Hu, Carbon, 2013, 59, 418–429 CrossRef CAS PubMed.
- X. Wang, M. Wu, W. R. Tang, Y. Zhu, L. W. Wang, Q. J. Wang, P. G. He and Y. Z. Fang, J. Electroanal. Chem., 2013, 695, 10–16 CrossRef CAS PubMed.
- Y. B. Zeng, Y. Zhou, L. Kong, T. S. Zhou and G. Y. Shi, Biosens. Bioelectron., 2013, 45, 25–33 CrossRef CAS PubMed.
- X. Yang, C. L. Chen, J. X. Li, G. X. Zhao, X. M. Ren and X. K. Wang, RSC Adv., 2012, 2, 8821–8826 RSC.
- Y. J. Guo, S. J. Guo, J. T. Ren, Y. M. Zhai, S. J. Dong and E. K. Wang, ACS Nano, 2010, 4, 4001–4010 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. W. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- H. K. He and C. Gao, Sci. China: Chem., 2011, 54, 397–404 CrossRef CAS.
- C. H. Xu, J. C. Wang, L. Wan, J. J. Lin and X. B. Wang, J. Mater. Chem., 2011, 21, 10463–10471 RSC.
- M. J. Lv, X. B. Wang, J. Li, X. Y. Yang, C. A. Zhang, J. Yang and H. Hu, Electrochim. Acta, 2013, 108, 412–420 CrossRef CAS PubMed.
- F. Gao, X. L. Cai, X. Wang, C. Gao, S. L. Liu, F. Gao and Q. X. Wang, Sens. Actuators, B, 2013, 186, 380–387 CrossRef CAS PubMed.
- X. Y. Xiao, P. R. Miller, R. J. Narayan, S. M. Brozik, D. R. Wheeler, I. Brener, J. Wang, D. B. Burckel and R. Polsky, Electroanalysis, 2014, 26, 52–56 CrossRef CAS.
- W. C. Zhu, T. Chen, X. M. Ma, H. Y. Ma and S. H. Chen, Colloids Surf., B, 2013, 111, 321–326 CrossRef CAS PubMed.
- J. Yan, S. Liu, Z. Q. Zhang, G. W. He, P. Zhou, H. Y. Liang, L. L. Tian, X. M. Zhou and H. J. Jiang, Colloids
Surf., B, 2013, 111, 392–397 CrossRef CAS PubMed.
- A. K. Yang, Y. Xue, Y. Zhang, X. F. Zhang, H. Zhao, X. J. Li, Y. J. He and Z. B. Yuan, J. Mater. Chem. B, 2013, 1, 1804–1811 RSC.
- M. L. Liu, Q. Chen, C. L. Lai, Y. Y. Zhang, J. H. Deng, H. T. Li and S. Z. Yao, Biosens. Bioelectron., 2013, 48, 75–81 CrossRef CAS PubMed.
- L. Wu, L. Y. Feng, J. S. Ren and X. G. Qu, Biosens. Bioelectron., 2012, 34, 57–62 CrossRef CAS PubMed.
- J. C. Wang, X. B. Wang, L. Wan, Y. K. Yang and S. M. Wang, Chin. J. Chem., 2010, 28, 1935–1940 CrossRef CAS.
- J. X. Chen and H. T. Jung, J. Phys. Chem. C, 2010, 114, 8227–8234 Search PubMed.
- D. C. Wei, Y. Q. Liu, Y. Wang, H. L. Zhang and L. P. Huang, Nano Lett., 2009, 9, 1752–1758 CrossRef CAS PubMed.
- Z. H. Sheng, L. Shao, J. J. Chen, W. J. Bao, F. B. Wang and X. H. Xia, ACS Nano, 2011, 5, 4350–4358 CrossRef CAS PubMed.
- C. X. Guo, Y. Lei and C. M. Li, Electroanalysis, 2011, 23, 885–893 CrossRef CAS.
- L. Tan, K. G. Zhou, Y. H. Zhang, H. X. Wang, X. D. Wang, Y. F. Guo and H. L. Zhang, Electrochem. Commun., 2010, 12, 557–560 CrossRef CAS PubMed.
- S. J. Li, J. Z. He, M. J. Zhang, R. X. Zhang and X. L. Lv, Electrochim. Acta, 2013, 102, 58–65 CrossRef CAS PubMed.
- Y. Y. Ling, Q. A. Huang, M. S. Zhu, D. X. Feng, X. Z. Li and Y. Wei, J. Electroanal. Chem., 2013, 693, 9–15 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 










