
Influence of charge compensating cations on propane adsorption in X zeolites: experimental measurement and mathematical modeling
Abstract
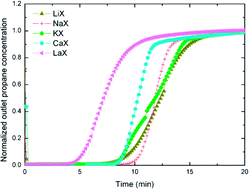
Separation of minor components is necessary prior to natural gas liquefaction. There are many methods to achieve this but one that has not been studied in great detail is adsorption of hydrocarbon gases on zeolite materials. A more comprehensive understanding of the fundamentals of hydrocarbon adsorption on zeolites is required in order to determine the efficacy of these materials in natural gas processing. This study investigates the influence of the charge compensating (non-framework) cation on the adsorption of propane on X zeolite by both dynamic experiments and mathematical modeling. This work presents a systematic experimental study examining the effects of the 5 typical types of charge compensating cations (Li+, Na+, K+, Ca2+, La3+) in X zeolites for saturated hydrocarbon adsorption. The dynamic experimental results reveal that for the X zeolites examined, all exhibited an affinity for propane, with LiX being the best, having a propane adsorption capacity of 15.5 wt%. Interestingly, unlike many non-zeolite solid sorbent materials, such as carbons, surface area and pore size alone do not necessarily determine propane adsorption capacity in these X zeolites. It has been shown that the charge compensating cation of the X zeolites of interest, in particular its valence, number of ions and size are the major factors affecting the propane adsorption capacity. Mathematical modeling equations are established by using mass balance in the adsorbent column, macroporous pellets and microporous crystals. The model-predicted results show a good match with our experimental results. The prediction results show that in our current experimental conditions, LiX has a slower adsorption rate than the other zeolites. The obtained adsorption equilibrium constants for all the X zeolites follow the same trend as their propane adsorption capacity, with LiX having the largest constant, suggesting a stronger binding energy between LiX and propane compared to the other zeolites.
 Please wait while we load your content...
Please wait while we load your content...

