On the kinetics and reaction mechanisms of boronic acid in interaction with diols for non-enzymatic glucose monitoring applications: a hybrid DFT study
Hadieh Monajemi*a,
Mun Hon Cheahb,
Vannajan Sanghiran Leeb,
Sharifuddin Mohd. Zainb and
Wan Ahmad Tajuddin Wan Abdullaha
aDepartment of Physics, Universiti Malaya, 50603 Kuala Lumpur, Malaysia. E-mail: h.monajemi@hotmail.com
bDepartment of Chemistry, Universiti Malaya, 50603 Kuala Lumpur, Malaysia
First published on 3rd January 2014
Abstract
Boronic acids are well-known for their ability to complex with saccharides and to form cyclic boronic esters or cyclic boronate ions. Increasing the reactivity of boronic acids would lead towards the design of a highly responsive sensor for non-enzymatic glucose monitoring applications. Many studies have been carried out to investigate the reactivity of boronic acids towards diols in different environmental effects. The symmetry around boron in their reactive species, however, is still an open question. In this study, we used computational quantum chemistry calculations to propose a model in which boronic acid is highly reactive towards diols. B3LYP/6-31+G(d,p) model chemistry was used in water to calculate the transition states of several proposed reaction mechanisms and the rates of the reactions were calculated using the transition state theory. The reaction takes place in two steps with the first step known as the rate determining step. The model we proposed in this study includes two different electronegative R-groups attached to boronic acid (i.e. CF3 and CH3) which highly influence the interaction of boron with diols. The kinetic results in our study show that boronate ion with a low electronegative R-group is a better sensor in alkaline medium and boronic acid with a high electronegative R-group is a better sensor in acidic medium. Using the latter in alkaline medium results in a very poor sensor since it is highly reactive and promptly forms a tetrahedral boronate ion. The high electronegative R-group on boronate ion decreases its basic characteristics and results in a low reactive sensor. The same scenario goes for the former i.e. boronate ion attached to a low electronegative R-group in acidic medium. As a result, we can conclude that boronic acid's reactivity is not mainly due to the symmetry around boron, it is due to the characteristics of boron itself which can be affected by both the R-group and the medium.
1 Introduction
Boronic acids are alkyl or aryl substituted boric acids which have a relatively stable low-polar C–B bond. They are well-known for their extensive applicability in the field of biomedicine.1 Among their charactericstics their ability to bind diol containing compounds such as saccharides would make them good sensors for measuring the concentration of biologically important sugars. Being covalent-based receptors, boronic acid sensors are superior to the synthetic receptors which function via hydrogen bonding.2 They rapidly and reversibly form cyclic boronic esters with diols through covalent bonds. Even though this characteristic of boric acids has been known for quite a long time,3 it was first observed in phenylboronic acid in 1954 upon its addition to a solution of saturated mannitol which eventually led towards the formation of cyclic boronic esters.4 This important finding was followed by several studies on the properties of boronic acids, of particular importance is the Lorand and Edwards' study in 1959 on the structure of benzeneboronate ion.5 It is well understood from Raman spectra that the structure of borate ion is similar to that of fluoborate due to the symmetry around boron. However, since boronic acids lack this symmetry around boron, it was still not known whether the conjugate basic form of boronic acid was trigonal (eqn (1)) or tetrahedral (eqn (2)).6
 | (1) |
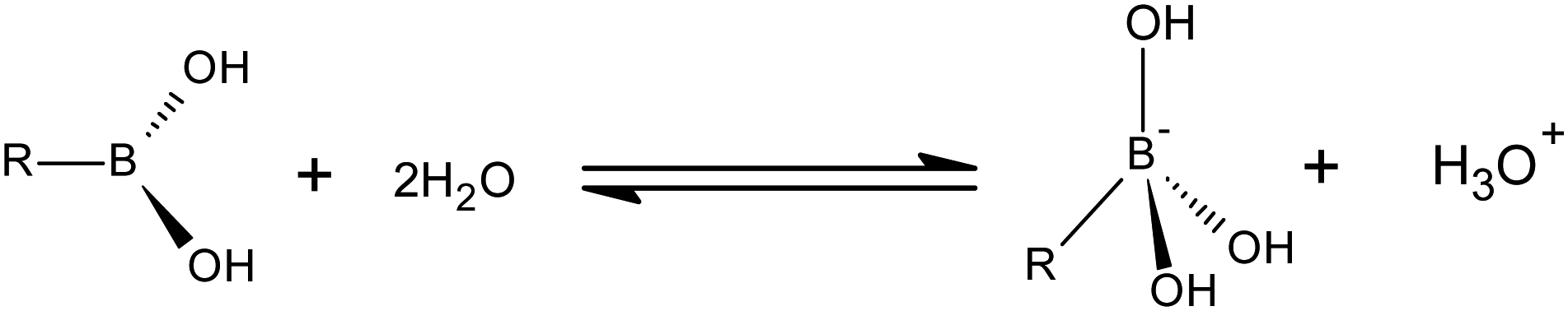 | (2) |
The method Lorand and Edwards used in their study involved the measurement of the equilibrium constants for the formation of complexes between benzeneboronate ion and hydroxyl containing compounds. Comparing these constants with that of borate, they concluded that the conjugate basic form of boronic acid and the boronate ester is tetrahedral. This resulted in a disagreement on what is the reactive form of boronic acid. It was suggested by Lorand and Edwards that boronate ion is reactive in alkaline medium since its complex formation lowers the pH. Several other studies have also suggested that the tetrahedral boronate ion is predominant since most of the reactions occur in alkaline solutions where sensors are most operative.7–12 On the other hand, there are a few arguments claiming that trigonal boronic acid is the reactive species regardless of the pH of the medium.13–18 The latter group has argued that the high reactivity of boronate ion in alkaline solution in which it exists abundantly is only due to the low concentration of its conjugate acid and does not necessarily mean that the basic boronate ion itself is more reactive than its conjugate acid. Their study shows that “trigonal boronic acid is always a reactive species irrespective of the pH of the solution and the reactivity of its conjugate tetrahedral boronate ion is comparable with or less than that of the boronic acid”.16,18,19 They have also recently validated their conclusion by setting up reaction systems using sensors and ligands with different pKa values, without proton ambiguity and a fully protonated ligand.19 They argue that even in alkaline solution, boronic acid is more reactive than its conjugate basic counterpart.
Determining the reactive species of boronic acid is an important step towards understanding its reaction mechanism in ester formation with diols. Another factor affecting its reactivity is the type of R-group attached to boron as this affects its sensitivity towards saccharides. The majority of studies which have been carried out have used either phenylboronic acid9,11 or boric acid.7,10,13,14 These studies have mainly focused on various combinations of diols and pH values to investigate the selectivity. We on the other hand, have focused on the R-group variety to investigate the reactivity of boronic acid. One way to look at this problem is to study the rate at which boronic acid or boronate ion reacts with diols. There are several limitations in experimental studies to figure out the accurate reaction mechanism and kinetic measurements because of the so called proton ambiguity; the transition structure cannot be clearly identified and the reaction of boronic acid in complexing diols can hardly be measured kinetically.8,14,16–18
In this study, a number of alternative reaction pathways between boronic acid and 1,2-ethanediol have been explored using Density Functional Theory (DFT) calculations. To explore the influence of the R group towards the reactivity of boronic acid, methylboronic acid (CH3B(OH)2) and trifluoromethylboronic acid (CF3B(OH)2) were selected because of the large difference in electronegativity.
2 Methodology
All the calculations were performed using Gaussian 09 software package.20 The structures of the reactants, transition state and products were optimized using B3LYP/6-31+G(d,p) model chemistry at a temperature of 298.15 K and a pressure of 1 atmosphere. The calculations were carried out in water using the integral equation formalism for the polarisable continuum model (IEFPCM).21 Vibrational frequencies were calculated to characterize the stationary points and zero point vibrational energies (ZPVE). The stationary points on the potential energy surface (PES) were categorized as minima with no imaginary frequency while the saddle point on the PES was known as the transition state with one imaginary frequency. The search for the quadratic region around the transition state was performed using the Quadratic Synchronous Transit (QST) approach and the final optimization of the transition structure was carried out using the Quasi-Newton algorithm. This method is known as the Synchronous Transit Quasi-Newton (STQN) method.22 The rate of reaction was calculated using the Eyring–Polanyi equation,
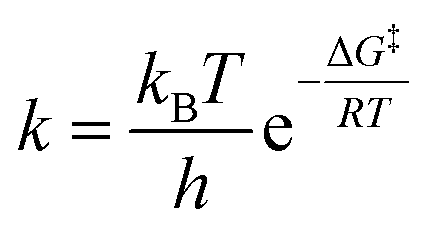 | (3) |
While higher levels of theory will produce more accurate energies, the purpose of this study was to discriminate between alternative reaction pathways by comparing the relative energy difference. The relative energy differences between CCSD (Coupled Cluster Single and Double excitation) and B3LYP (Becke three parameter hybrid functional combined with Lee–Yang–Parr correlation functional) methods calculated for the first proposed reaction mechanism in this study are presented in Table 1. A comparison was made of the relative activation energies determined by CCSD and B3LYP methods for the reaction of CH3B(OH)2 and CF3B(OH)2 with diol. It was observed that the DFT method follows the same trend as the coupled cluster method and there is little variation between the two methods. Furthermore, DFT methods have been shown to reproduce fairly high accuracy.24,25 Due to the relatively high computational cost associated with CCSD, DFT was adopted for the purpose of this study.26–29
| Structure | Method/basis set | ΔG‡ (kcal mol−1) | k s−1 |
|---|---|---|---|
| CH3B(OH)2 | CCSD/aug-cc-pVDZ (PCM) | 37.652 | 2.338 × 10−15 |
| B3LYP/6-31+G(d,p) (PCM) | 38.110 | 1.078 × 10−15 | |
| CF3B(OH)2 | CCSD/aug-cc-pVDZ (PCM) | 28.866 | 6.555 × 10−9 |
| B3LYP/6-31+G(d,p) (PCM) | 29.091 | 4.481 × 10−9 |
The basis function we used describes the set of d-functions, added to the heavy atoms as well as a set of p-functions, added to the hydrogen atoms (i.e. d,p). This function is referred to as a polarization function. We also included the diffuse function, representing the portion of atomic orbitals which are distant from the nuclei. The diffuse functions that are denoted by a “+” sign, are particularly important for the anionic structures. In addition for heavy atoms, we also added the diffuse function to light atoms such as hydrogen in the coupled cluster method, indicated by an “aug” sign for the CCSD method.
Five possible reaction paths were considered using protonated and deprotonated forms of both boronic acid and 1,2-ethanediol (mechanisms I–V) leading to two distinct products (products 1 and 2).
3 Results and discussion
Boronic acids are known to react with diol compounds to form cyclic esters.3 Kinetic studies thus far have suggested a two step mechanism for the formation of a cyclic ester where the rate determining step is often attributed to initial binding of the diol to the boron.10,14 A large negative value of the activation entropy during the first step of the reaction has been reported which indicates an increase in the coordination number of the central boron atom.7,8,13 However it should be noted that there are reports of ring closure being the rate determining step due to steric strain.30 In this study we explored the reactivity of neutral Lewis boronic acid (mechanism I and II) as well as Lewis (mechanisms III) and Brønsted (mechanisms IV and V) basic boronate ions (Fig. 1 and 2). Two different R-groups with a large difference in electronegativity were used in this study i.e. CH3–B(OH)2 and CF3–B(OH)2. | ||
| Fig. 1 Five different reaction mechanisms for methylboronic acid in interaction with 1,2-ethanediol in water. All reactions take place in two steps, forming an intermediate in the middle. | ||
 | ||
| Fig. 2 Five different reaction mechanisms for trifluoromethylboronic acid in interaction with 1,2-ethanediol in water. All reactions take place in two steps, forming an intermediate in the middle. | ||
3.1 Proposed reaction mechanisms
Five possible reaction pathways for the reaction between the trigonal forms of R–B(OH)2 and 1,2-ethanediol were explicitly considered as indicated in Fig. 1 and 2.Mechanism I proposes a reaction between the neutral diol and neutral boronic acid. The empty p orbital on the boron atom deshields its nucleus which increases its Lewis acidity. This would result in an interaction between the boron atom in boronic acid and the electron lone pair on the oxygen of the incoming diol.
Although a few studies have shown that the complexation reaction of tetrahedral boronate ion is 3–4 orders of magnitude faster than that of trigonal boronic acid,8–10 it is not wise to conclude that the tetrahedral boronate ion is the reactive species since it would be rather difficult to specify the rate determining step in a two step mechanism. The best way to rationalize this idea is to propose a reaction mechanism in which the trigonal boronic acid in reaction with a diol forms a tetrahedral intermediate. Based on what has been reported above and since the chelation reaction is not the rate determining step, it is more likely for the trigonal boronic acid to be the starting material. This could occur with either a Brønsted basic sensor or a basic incoming ligand. In the former, the diol donates its proton to react with trigonal boronic acid and forms a tetrahedral intermediate (mechanisms IV and V). The latter occurs when the incoming ligand lacks an acidic proton: the negative oxygen tends to react with the Lewis acidic boron (mechanisms II and III). This reaction mechanism could explain the idea pointed out by Pizer and Tihal, in which for a ligand lacking an acidic proton, trigonal boronic acid would be unreactive.8,9 This is only true if the intermediate is trigonal with elimination of a water molecule by removal of hydroxide from the central boron and a proton from the ligand (mechanism II). Otherwise, one proton is enough for the whole reaction to occur with the negative oxygen of the ligand directly attacking the boron atom, forming a tetrahedral intermediate which could easily occur if the starting material is in the trigonal form (mechanism III).
Furthermore, for ligands with no acidic proton, an unchelated tetrahedral intermediate would still form, increasing the coordination number of boron from 3 (sp2) to 4 (sp3). The result of this reaction is a tetrahedral intermediate in which the negatively charged boronate ion has a low first ionization energy. In addition, with electronegative oxygen attached to boron, the B–O bond becomes relatively polar and less stable than that for trigonal boronic acid. Thus, an acidic environment could facilitate the chelate ring closure due to a good leaving hydroxyl group. In this proposed mechanism, a lack of one acidic proton does not have a negative income on the reactivity of boronic acid and makes it rather efficient.
3.2 Reaction kinetics for the reaction of CH3–B(OH)2 and CH3–BOOH− with 1,2-ethanediol
The kinetic data were analyzed based on the reaction mechanisms presented in Fig. 1. A relatively high activation barrier was observed in the first step of mechanism I which is known to be the rate determining step, resulting in a rate of 10−15 s−1 (Table 2). The rate of reaction was enhanced by a factor of 107-fold during the chelate ring closure which is in good agreement with previous experiments.8,10,14 An example of such a study is the one carried out by Pizer and Tihal (1992) in which the rate of reaction between methylboronic acid and polyols in the second step was 102-fold higher than that of the first step. This mechanism ultimately leads to a trigonal cyclic boronic ester with elimination of two water molecules. The increased rate during chelate ring closure could be due to the water molecule interacting with the intermediate and acting as an anchor to stabilize the transition structure. The rate enhancement of the chelate ring closure in mechanism I was however not as great as that observed for mechanism II in which the incoming diol lacks an acidic proton. In addition to the eliminated water molecule, the negative charge on the hydroxyl of diol further stabilizes the transition structure, increasing the rate by a factor of 1023-fold for CH3–B(OH)2 (Fig. 1 and Table 2). Despite the high rate during chelate ring closure, mechanism II is very unlikely to happen due to a very high activation barrier during the first step of the reaction. This is in good agreement with the highlighted lower reactivity of HL− compared to that of H2L (H2L being the bidentate ligand) towards boronic acid.19| Mechanism/CH3B(OH)2 | Step of reaction | ΔG (kcal mol−1) | ΔS (kcal mol−1 K−1) | ΔG‡ (kcal mol−1) | ΔS‡ (kcal mol−1 K−1) | k s−1 |
|---|---|---|---|---|---|---|
| I/neutral | Step 1 | 6.621 | −0.031 | 38.110 | −0.047 | 1.078 × 10−15 |
| Step 2 | −4.261 | 0.014 | 28.123 | −0.010 | 2.300 × 10−8 | |
| Overall | 2.401 | −0.017 | — | — | 1.078 × 10−15 | |
| II/acidic | Step 1 | 0.549 | −0.036 | 47.785 | −0.050 | 8.566 × 10−23 |
| Step 2 | −8.210 | −0.007 | 17.625 | −0.006 | 1.163 | |
| Overall | −7.661 | −0.043 | — | — | 8.566 × 10−23 | |
| III/acidic & basic | Step 1 | −4.273 | 0.042 | — | — | — |
| Step 2 | −3.396 | −0.001 | 8.035 | 0.003 | 1.269 × 107 | |
| Overall | −7.661 | −0.043 | — | — | 1.269 × 107 | |
| IV/basic | Step 1 | −12.208 | −0.044 | 20.254 | −0.042 | 1.369 × 10−2 |
| Step 2 | −3.396 | −0.001 | 8.035 | 0.003 | 1.269 × 107 | |
| Overall | −15.590 | −0.045 | — | — | 1.369 × 10−2 | |
| V/basic | Step 1 | −3.081 | −0.032 | 34.819 | −0.047 | 2.805 × 10−13 |
| Step 2 | 0.001 | −0.012 | 0.755 | −0.003 | 0.279 × 1013 | |
| Overall | −3.071 | −0.044 | — | — | 2.805 × 10−13 |
A few other experimental studies have also reported a decrease in the reactivity of boronic acid as the ligand deprotonates.7,14 Ishihara et al. (1991) reported a lower rate by a factor of 104-fold for 4-isopropyltropolone (Hipt) compared to chromotropic acid (H2cht) as the ligand and Ito et al. (2003) reported a decrease in the reactivity of boronic acid as H2ipt+ deprotonates to Hipt and ipt−. However, both their proposed mechanisms, H2cht in Ishihara et al.'s study and H2ipt+ in Ito et al.'s study result in tetrahedral boronate ion and elimination of a hydronium ion which is different from mechanism I in our study. It is worth mentioning that their studies were carried out in alkaline solution where boronic acid predominantly exists in its basic form and is more reactive with fully protonated diols.
In mechanism III on the other hand, having a deprotonated ligand is an advantage since the first step goes through no activation barrier and the reaction takes place at a rather high rate, forming a tetrahedral intermediate. However, the rate of formation of cyclic boronate ester in mechanism III with a tetrahedral boronate ion as the starting material is rather high compared to the other steps. This could support the idea of a faster reaction rate for tetrahedral boronate ion compared to that for trigonal boronic acid.
Mechanisms IV and V represent the reaction in alkaline medium where boronic acid is present in a trigonal basic form. The second step of mechanism V is similar to the first step of mechanism IV where the proton from the ligand is transferred to the basic oxygen of boronate ion. The rates are however significantly different knowing that the former yields a tetrahedral cyclic boronate ester and the latter yields a Lewis basic tetrahedral intermediate. This indicates the low stability of the trigonal basic form of boronic acid and its tendency to form a tetrahedral Lewis base which further supports the idea of tetrahedral boronate ion as the conjugate basic form of boronic acid. The overall rates of reaction in these mechanisms are relatively high which indicates the importance of a fully protonated ligand for boronate ions.
3.3 Reaction kinetics for the reaction of CF3–B(OH)2 and CF3–BOOH− with 1,2-ethanediol
In this section, we elaborate the reactivity of CF3–B(OH)2 based on the mechanisms presented in Fig. 2. The activation free energy and rate of reaction for all the proposed mechanisms are presented in Table 3. The data presented for mechanism I show that for a high electronegative R-group, the rate of reaction is increased by a factor of 106 and 10-fold in the first step and second step respectively. The transition structures in the rate determining step for both R-groups are however identical with similar activation entropies. On the other hand, the transition structure in step 2 of mechanism I is different from that for CH3B(OH)2. The slightly higher activation entropy explains the lower activation barrier and higher reaction rate for this mechanism.| Mechanism/CF3B(OH)2 | Step of reaction | ΔG (kcal mol−1) | ΔS (kcal mol−1 K−1) | ΔG‡ (kcal mol−1) | ΔS‡ (kcal mol−1 K−1) | k s−1 |
|---|---|---|---|---|---|---|
| I/neutral | Step 1 | 4.890 | −0.030 | 29.091 | −0.047 | 4.481 × 10−9 |
| Step 2 | 0.411 | 0.016 | 26.862 | −0.015 | 1.937 × 10−7 | |
| Overall | 5.301 | −0.014 | — | — | 4.481 × 10−9 | |
| II/acidic | Step 1 | −4.780 | −0.035 | 32.161 | −0.039 | 2.503 × 10−11 |
| Step 2 | −24.160 | 0.001 | 9.601 | −0.003 | 0.900 × 106 | |
| Overall | −28.940 | −0.040 | — | — | 2.503 × 10−11 | |
| III/acidic & basic | Step 1 | −25.601 | −0.037 | — | — | — |
| Step 2 | −3.330 | −0.003 | 26.640 | −0.009 | 2.819 × 10−7 | |
| Overall | −28.940 | −0.040 | — | — | 2.819 × 10−7 | |
| IV/basic | Step 1 | −5.630 | −0.035 | 24.141 | −0.037 | 1.923 × 10−5 |
| Step 2 | −3.330 | −0.003 | 26.640 | −0.009 | 2.819 × 10−7 | |
| Overall | −8.960 | −0.038 | — | — | 2.819 × 10−7 | |
| V/basic | Step 1 | 4.230 | −0.024 | 36.120 | −0.042 | 3.113 × 10−14 |
| Step 2 | −15.010 | −0.007 | 19.460 | −0.011 | 5.238 × 10−2 | |
| Overall | −10.780 | −0.032 | — | — | 3.113 × 10−14 |
As for mechanism I, the rate also increased in mechanism II where the diol lacks an acidic proton. The electronegative R-group in this structure slightly increases the acidity of boron and the electrophilicity which increases its tendency to react with the nucleophilic oxygen of diol. This effect is observed more obviously in the second step of mechanism II with a relatively high rate (106 s−1) where the chelate ring closure does not involve deprotonation of the diol since it already lacks a proton. Furthermore, the higher electronegative R-group of CF3–B(OH)2 results in a 105-fold higher rate in this step compared to that for CH3–B(OH)2.
Mechanism III is less likely to occur compared to that for CH3–B(OH)2 due to a relatively high activation barrier in the second step. In this mechanism, the tetrahedral intermediate is in the Lewis base form which makes boron a high electron donor. The high electronegative R-group lowers this characteristic and results in a less electron donating boron which decreases the rate of reaction by a factor of 1014-fold compared to that for CH3–B(OH)2. The same effect was also observed for the Brønsted basic form of boronic acid which lacks a proton on one of the OH groups. The high electronegativity reduces the basic characteristic of boron, resulting in a 103-fold and 10-fold decrease in step 1 of mechanisms IV and V respectively. The second step of mechanism IV is similar to that for mechanism III. However, it is the rate determining step for mechanism IV with CF3 R-group. The higher activation barrier reduces the rate by a factor of 10−2-fold relative to step 1 of mechanism IV.
The effect of the electronegative R-group was further observed in mechanism V where both steps have a relatively higher activation barrier compared to that for the CH3 R-group. Although the TS2V seems rather stable due to a simple proton transfer between the ligand and the basic oxygen of boron and indeed it is for CH3 R-group, it requires a fairly high activation energy with the CF3 R-group to proceed with the chelate ring closure. It is the electronegative R-group which reduces the stability of the transition structure and affects the reaction rate. These results are consistent with those carried out empirically by Watanabe et al.19 Among the R-groups they used were 3-fluorophenylboronic acid (3-FPhB(OH)2) and 3-pyridylboronic acid (3-pyB(OH)2) with the former being more electronegative. The reaction rate for tetrahedral 3-fluorophenylboronate ion is 104-fold lower than that for 3-pyridylboronate ion. Furthermore, the rate of reaction is 103-fold higher for 3-FPhB(OH)2 compared to its tetrahedral Brønsted basic form (i.e. 3-FPhB(OH)3−).
3.4 Comparing the reactivity of CH3–B(OH)2 and CF3–B(OH)2 towards 1,2-ethanediol
The effect of an electronegative R-group on the reactivity of boronic acid was clearly observed in all the reaction mechanisms. However, a higher electronegativity only increases boronic acid's reactivity in its Lewis acidic form (mechanisms I and II); the rate was enhanced by a factor of 106-fold and 10-fold in the first and second step of mechanism I and 1012-fold and 106-fold in the first and second step of mechanism II respectively. Among all these mechanisms with negatively charged sensor or ligand, mechanisms I and II are the most unfavorable ones. On the other hand, mechanism III turns out to be rather favorable with an electropositive R-group. The nucleophilic attack of a negatively charged oxygen with boron in mechanism III yields a tetrahedral intermediate which has a Lewis basic characteristic. For this mechanism, the chelate ring closure is similar to the second step of mechanism I which involves water elimination. The rate of this mechanism is 1014-fold higher for CH3–B(OH)2. This can be rationalized by considering the higher electronegativity of CF3 which reduces the Lewis basicity of boronate ion, resulting in a higher activation barrier. Furthermore, a higher electronegative R-group enhances the electrophilicity of boron and results in a stronger B–O bond, hence a poorer OH leaving group.Associated with this is the fact that the more electronegative CF3-group also reduces the basicity of the Brønsted boronate ion (Tables 2 and 3). This results in less nucleophilic basic oxygen on boronic acid which causes a delay in the proton transfer of incoming diol to the boronic acid leaving group. The difference was clearly observed in TS1IV, TS2III&IV, TS1V and TS2V. There was a 103-fold reduction in the reaction rate upon using a CF3 R-group rather than a CH3 R-group in the first step of mechanism IV.
In mechanism V, the neutral diol reacts with boron in a similar fashion as in the first step of mechanism I which also results in a lower rate by a factor of 10-fold for the CF3 R-group. The second step involves a proton transfer from the other end of the diol followed by nucleophilic attack of the resulting negatively charged oxygen towards boron which leads to the formation of a tetrahedral cyclic boronate ester (product 2). Similar to the first step of mechanism IV, the second step of mechanism V involves a proton transfer from the hydroxyl of diol to the Brønsted basic oxygen of boronate ion. The resulting negative oxygen of diol becomes highly reactive with a high tendency to form a tetrahedral Lewis boronate ion. This is clearly understood from the significantly lower activation barrier in mechanism V with a lower electronegative R-group (Fig. 1).
This study made it rather clear that regardless of the properties of the incoming ligand, boronate ions always have Lewis characteristics in forming the cyclic boronate ester. Lewis bases eliminate a water molecule and form a tetrahedral cyclic boronate ester. Brønsted bases initially form a Lewis acid by accepting a proton from the ligand and then the negatively charged oxygen on the ligand forms a tetrahedral intermediate with electrophilic boron on the boronic acid which ultimately leads to the formation of a cyclic boronate ester. For bifunctional ligands lacking an acidic proton, trigonal boronic acid is more reactive due to the empty p orbital on boron; the nucleophilic oxygen of the ligand attacks the electrophilic boron and forms a tetrahedral intermediate without any leaving OH. Chelate ring closure then takes place by proton transfer from the ligand to the leaving OH.
We have evidently observed in this study that boronic acids could be highly reactive in both acidic and basic forms under the effect of different R-groups. An electronegative R-group enhances the reactivity of boronic acids while retarding the reactivity of boronate ions. Therefore, boronic acids attached to highly electronegative R-groups are more reactive in acidic environment, while boronate ions are more reactive in alkaline medium with highly electropositive R-groups. However, using boronic acid in alkaline medium with electronegative R-group results in a weak sensor since it promptly forms a tetrahedral boronate ion upon reacting with the bases in the medium. Now that the structure is basic, being attached to an electronegative R-group further decreases its basicity and as a result, its reactivity. On the other hand, a tetrahedral boronate ion attached to a lower electronegative R-group results in a good sensor due to the fact that boron's basic characteristic and as a result, its reactivity is enhanced. The same scenario is also true for trigonal boronic acids attached to highly electronegative R-groups. Being highly reactive in alkaline medium, they make a better sensor in acidic environment.
4 Conclusion
The reactivity of boronic acid is based on many factors including the R-group, the incoming diol and the medium. The data in this study indicate that various R-groups can affect the reactivity of boronic acid based on the pKa of the ligand and sensor as well as the pH of the system. In the case of alkaline solution, a highly electronegative R-group results in a highly reactive boronic acid at first which promptly turns into a boronate ion. Thus, it is ultimately turned into a basic sensor attached to a highly electronegative R-group which is shown in this study to decrease the reactivity of boronic acid by decreasing boron's basicity. Thus, due to the fact that the sensors function better in alkaline media, this study suggests that using a lower electronegative R-group and even an electropositive one attached to a basic boronate ion increases the sensor's sensitivity towards diols. Furthermore, from a comparison of different reaction mechanisms and rates of reaction, one can conclude that the boronate ion is more stable in the form of a Lewis base in alkaline medium and therefore, it predominantly exists in the tetrahedral form rather than the trigonal form.Acknowledgements
This research is supported by High Impact Research Grant UM-MOE UM.C/625/1/HIR/MOE/F00004-21001 from the Ministry of Education Malaysia. This research is also conducted with the support of a University of Malaya Research Grant (UMRG-Project no. RP001C-13ICT)/Computation & Informatics (C + i) Research Cluster/High-Performance Scientific Computing Program from the University of Malaya (UM) and the Institute of Research Management and Consultancy of University of Malaya (PPP) with grant number PS018-2012A.References
- J. N. Cambre and B. S. Sumerlin, Polymer, 2011, 52, 4631–4643 CrossRef CAS PubMed.
- A. P. Davis and R. S. Wareham, Angew. Chem., Int. Ed., 1999, 38, 2978–2996 CrossRef , It is due to the fact that no hydrogen-bonding based receptor has yet been designed to compete with bulk water in aqueous media for low concentrations of saccharides, It is due to the fact that no hydrogen-bonding based receptor has yet been designed to compete with bulk water in aqueous media for low concentrations of saccharides.
- J. Böeseken, Adv. Carbohydr. Chem., 4, 189–210 Search PubMed , The absolute structural configuration of hydroxyl-containing compounds, more importantly saccharides was determined by Böeseken and co-workers using the known ability of boric acids in complexing diols. Upon addition of the saccharides to the solution of boric acid, an increase in both acidity and conductivity was observed. By tracking those changes, they were able to figure out the relative configuration of the hydroxyl groups.
- H. G. Kuivila, A. H. Keough and E. J. Soboczenski, J. Org. Chem., 1954, 19, 780–783 CrossRef CAS.
- J. P. Lorand and J. O. Edwards, J. Org. Chem., 1959, 24, 769–774 CrossRef CAS.
- J. O. Edwards, G. C. Morrison, V. F. Ross and J. W. Schultz, J. Am. Chem. Soc., 1955, 77, 266–268 CrossRef CAS , The symmetry around boron in boric acid structure results in analogous behaviour of its conjugate base to fluoborate. Therefore, boric acid's conjugate basis form is known to be tetrahedral.
- K. Ishihara, Y. Mouri, S. Funahashi and M. Tanaka, Inorg. Chem., 1991, 30, 2356–2360 CrossRef CAS.
- R. Pizer and C. Tihal, Inorg. Chem., 1992, 31, 3243–3247 CrossRef CAS.
- R. Pizer and C. Tihal, Polyhedron, 1996, 15, 3411–3416 CrossRef CAS.
- C. Shao, S. Matsuoka, Y. Miyazaki, K. Yoshimura, T. M. Suzuki and D. A. P. Tanaka, J. Chem. Soc., Dalton Trans., 2000, 3136–3142 RSC.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- S. Shinkai and M. Takeuchi, Biosens. Bioelectron., 2004, 20, 1250–1259 CrossRef CAS PubMed.
- S. Kagawa, K.-I. Sugimoto and S. Funahashi, Inorg. Chim. Acta, 1995, 231, 115–119 CrossRef CAS.
- H. Ito, Y. Kono, A. Machida, Y. Mitsumoto, K. Omori, N. Nakamura, Y. Kondo and K. Ishihara, Inorg. Chim. Acta, 2003, 344, 28–36 CrossRef CAS.
- T. Matsumura, S. Iwatsuki and K. Ishihara, Inorg. Chem. Commun., 2005, 8, 713–716 CrossRef CAS PubMed.
- S. Iwatsuki, S. Nakajima, M. Inamo, H. D. Takagi and K. Ishihara, Inorg. Chem., 2007, 46, 354–356 CrossRef CAS PubMed.
- S. Iwatsuki, Y. Kanamitsu, H. Ohara, E. Watanabe and K. Ishihara, J. Phys. Org. Chem., 2012, 25, 760–768 CrossRef CAS.
- C. Miyamoto, K. Suzuki, S. Iwatsuki, M. Inamo, H. D. Takagi and K. Ishihara, Inorg. Chem., 2008, 47, 1417–1419 CrossRef CAS PubMed.
- E. Watanabe, C. Miyamoto, A. Tanaka, K. Iizuka, S. Iwatsuki, M. Inamo, H. D. Takagi and K. Ishihara, Dalton Trans., 2013, 42, 8446–8453 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, H. P. H. X. Li, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford CT, 2010.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed , The basic concept in all continuum models is the cavity where the solute is placed in within a continuous dielectric medium, mimicking the solvent. The charge distribution of the solute inside the cavity and the dielectric continuum interact with one another in an iterative procedure which corresponds to the self consistent process. The size and shape of the cavity depends on the type of the continuum model. In particular it should exclude the solvent and its boundaries should contain the largest possible part of the solute charge distribution. The PCM models belong to the category of the ASC (apparent surface charge). One of the successive reformulations of the PCM model is the IEFPCM (integral equation formalism polarisable continuum model) in which the potentials are redefined in terms of a proper Green functions.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- H. Eyring, J. Chem. Phys., 1935, 3, 107–115 CrossRef CAS PubMed.
- L. Goerigk and S. Grimme, Phys. Chem. Chem. Phys., 2011, 13, 6670–6688 RSC.
- A. P. Scott and L. Radom, J. Phys. Chem., 1996, 100, 16502–16513 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter, 1992, 45, 13244–13249 CrossRef.
- Y. Yamamoto, T. Matsumura, N. Takao, H. Yamagishi, M. Takahashi, S. Iwatsuki and K. Ishihara, Inorg. Chim. Acta, 2005, 358, 3355–3361 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |