
Investigation of in situ annealing on poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate): towards all-solution-processed inverted polymer solar cells
Abstract
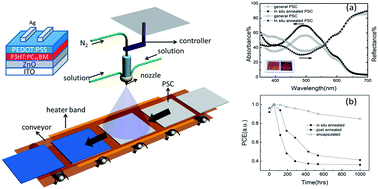
Efficient spray-coated inverted polymer solar cells (PSCs) based on a poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) hole transport layer (HTL) are investigated by incorporating an in situ annealing treatment. Compared to conventional annealed devices, the in situ annealed PSCs exhibited a 25.5% and 47.7% enhancement in fill factor and power conversion efficiency (PCE) respectively, as well as about a two-fold improvement in the device lifetime. The performance enhancement was attributed to the improvement of the vertical phase separation of the PEDOT:PSS HTL and the reduction of the residual humidity of the HTL. This approach significantly enhances our understanding of the application of the PEDOT:PSS HTL in inverted PSCs, and illuminates the potential of spray coating for the sustainable commercial fabrication of PSCs with high PCEs.
 Please wait while we load your content...
Please wait while we load your content...

