Aerosol-assisted deposition of gold nanoparticle-tin dioxide composite films†
Clair Chewa,
Peter Bishopb,
Carmen Salcianub,
Claire J. Carmalta and
Ivan P. Parkin*a
aMaterials Chemistry Research Centre, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H OAJ, UK. E-mail: i.p.parkin@ucl.ac.uk
bJohnson Matthey Research Center, Sonning Common, Reading, UK
First published on 26th February 2014
Abstract
Composite gold/tin oxide films were grown from a simple one-pot precursor solution containing monobutyltin trichloride and auric acid in methanol using aerosol assisted chemical vapour deposition (AACVD). Two types of films were formed on glass – gold nanoparticle films on the top plate and Au/SnO2 composite film on the heated substrate. The composite film showed different amounts of gold incorporation with position from the inlet – enabling a graded film to be formed. This enabled rapid assessment of the films' functional properties. Optimum locations showed a combination of desirable properties including a blue colouration and reflectivity in the IR. This work shows that the colour of TCO films can be tuned by gold nanoparticle dopants without removing the TCO properties.
1 Introduction
Nanometre sized gold nanoparticles exhibit strong surface plasmon resonance (SPR).1 The SPR can be seen by an absorption in the UV-visible spectra which arises when the incoming light is coherent with the electrons oscillating on the surface of the nanoparticle.1 This effect is exploited for uses such as in third order linear optics2 and surface enhanced Raman spectroscopy.3 Furthermore the incorporation of noble nanoparticles into metal oxides has been said to improve desirable properties such as the photocatalytic properties of TiO2,4 electrochromic performance of WO3 (ref. 5) and gas sensing sensitivities of SnO2.6 The strong SPR in the visible range of gold nanoparticles also enables them to be incorporated into glass during production process to make aesthetically pleasing body tinted coloured glasses – such as those used in stained glass windows.7 However, the nanoparticles do not need to be incorporated into the body of the glass to have an effect, they can be inserted into thin films used as window coatings. The colour of the nanocomposite films will depend on both the nanoparticles – the shape, size, nature as well as the surroundings. Therefore, the resulting colour of the film should be tunable according to the properties of the nanoparticles.Tin dioxide is a widely deposited transparent conducting oxides (TCO) in its F-doped form, especially for low emissivity architectural windows – it is highly reflective in the IR8 with a large band gap of 3.6 eV.9 These “smart” windows can have more advanced forms when combined with an electrochromic layer which is able to change colour when certain voltages are applied.10,11 In optoelectronic devices such as flat panel displays, touch screens and photovoltaic cells, tin oxide is utilised as a transparent electrode. Furthermore, tin oxide has also been found to be an effective detector for gas sensing devices.12,13 Tin oxide is favoured because of its high mechanical and thermal stability, making it long lasting and able to withstand high temperature processes.14 An insulator when stoichiometric, tin oxide becomes conducting when oxygen vacancies are generated in it or when doped with elements such as F or Sb.15 Even when tin oxide is in this n-type semiconductor state the band gap is still wide enough to maintain transparency.16 Although having slightly lower conductivity fluorine doped tin oxide (FTO) is a viable alternative to the industry leader indium – tin oxide, being cheaper and having higher thermal stability.17 Commercial FTO glass is mainly deposited by spray pyrolysis or by chemical vapour deposition (CVD) for example onto hot glass during float glass production.11,18
We report an investigation into the formation of tin oxide films with embedded gold nanoparticles by AACVD. Aerosol assisted CVD requires an aerosol to be generated from a precursor solution and deposited onto a hot substrate. The organic substances breaks down and leaves behind a thermally stable thin film often of desired composition.19 This process is easily reproducible and similar to industrial scale reactions. Many precursors have been investigated for the deposition of tin oxide,20 this paper uses monobutyl tintrichloride (MBTC) as a precursor. The type of CVD described here generates the precursor aerosol using an ultrasonic humidifier and is termed aerosol assisted CVD (AACVD).
One of the primary reasons for investigating this system was to see if gold could be used as colourant in a CVD derived film. Many methods exist for producing coloured glass, the main one is to add metals into the bulk of the glass giving body tinted glass.21 The disadvantage of this method is that on a commercial scale the glass holding tank can take weeks to change colour if a different coloured glass is required. By incorporating nanoparticles exceptional colouration efficiency within a thin film coating or glass would be an alternative method by which coloured glass could be made. This would not suffer from the change over problems of body tinted glass. The tuning of colour has been investigated with a nanocomposite VO2 film with Au nanoparticles.22 Using this AACVD method we can make Au nanocomposite SnO2 films in an adherent way suitable for us as part of an industrial glass manufacturing process.
The reaction conditions were designed so that we could generate gradients of both nanoparticle and tin dioxide on the film of different ratios. The reason for this was so that we could rapidly explore phase composition space from a limited number of deposition runs and by incorporating nanoparticles at different concentrations films with different colour should be obtainable. Furthermore the inclusion of metal nanoparticles would also be expected to effect the glass conductivity and reflectivity properties – perhaps enhancing their behaviour as a solar reflective coating.23
2 Experimental
2.1 Materials
N-Butyltin trichloride (monobutyltin trichloride or MBTC) was purchased from Sigma-Aldrich Chemical Co; methanol from Merck Chemicals and hydrogen tetrachloroaurate(III) hydrate was supplied from Alfa Aeser by Johnson Matthey PLC.2.2 Precursor solutions
Butyltin trichloride (2 mmol) and varying amounts of hydrogen tetrachloroaurate(III) hydrate were dissolved into 20 ml of methanol and left to stir thoroughly before being used in the AACVD reaction. The ratio of 0.06 Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sn was chosen to be studied in a variety of temperatures because it gave a good gradient range of gold depositions when compared to the others.
:Sn was chosen to be studied in a variety of temperatures because it gave a good gradient range of gold depositions when compared to the others.
2.3 Aerosol assisted chemical vapour deposition
Depositions were carried out in a cold-walled horizontal-bed CVD reactor. The reactor contained a top and bottom plate for deposition to occur, both composed of SiO2 barrier glass (dimensions: 145 × 45 × 5 mm) supplied by Pilkington NSG. Deposition was carried out on the barrier layer to prevent possible ion transfer from the bulk glass. A carbon block under the bottom plate heated the CVD reactor. The top plate was positioned 8 mm above and parallel to the bottom plate, the complete assembly was enclosed within a quartz tube. The aerosol of the precursor solution was generated using a PIFCO-HEALTH ultrasonic humidifier with an operating frequency of 50 kHz and 20 W of power. The aerosol generated was moved to the reactor using a nitrogen gas flow via polytetrafluoroethylene and glass tubing, where it entered through the inlet to the top and bottom plate. The reactor waste gas left through an exhaust. The flow of nitrogen carrier gas transported the vapour from the flask until all liquid was spent, this took typically 30–45 minutes. The flow of nitrogen was maintained at 2.0 l min−1. The temperature of the carbon heating block was varied between 400 and 500 °C with corresponding changes in film characteristics determined. The heat and nitrogen flow into the reactor was then turned off and left to cool to room temperature. The cooled plates were removed and handled in air.2.4 Film analysis
Scanning electron microscopy (SEM) was carried out on a JEOL 6301F instrument using voltages between 5 and 20 kV. Energy Dispersive X-ray (EDX) analysis was also carried out on the same instrument at an operating frequency of 20 kV. Powder X-ray Diffraction (XRD) patterns were measured on a Bruker-Axs D8 (GADDS) diffractometer using monochromated Cu Kα radiation. UV-vis spectra were taken using a Thermo Spectronic Helios Alpha single beam instrument over a range of 200–2500 nm. Conductance measurements were taken with an ammeter.2.5 Results of SnO2 and gold nanoparticle films
Films of SnO2 were deposited from a precursor solution of BuSnCl3 (2 mmol) in methanol (20 ml) at 400, 450 and 500 °C. The gold nanoparticle film was deposited at 450 °C from a solution of HAuCl4 (0.12 mmol) in methanol (20 ml). | ||
| Fig. 1 Schematic of an aerosol assisted chemical vapour deposition set up and regions where measurements were taken at 1 (a), 5 (b) and 9 cm (c) away from the inlet. | ||
 | ||
| Fig. 2 XRD patterns of SnO2 (left) deposited at 400, 450 and 500 °C and a typical XRD pattern observed the gold nanoparticle films. | ||
The gold depositions were investigated in SEM (Fig. 3) and it was observed that the films were discontinuous, having either particulate or island morphology. This indicates that the reduction of auric acid into bulk gold occurs in the precursor droplet prior to deposition otherwise it might be expected that if the auric acid was reduced on the surface of the glass, a thin film of gold should form.
 | ||
| Fig. 3 SEM images of auric acid showing island growth on the bottom plate (left) and nanoparticle deposition on the top plate. | ||
 | ||
| Fig. 4 Transmittance (a) and reflectance (b) spectra of SnO2 deposited at different temperatures. | ||
 | ||
| Fig. 5 Transmittance, reflectance (a) and absorption (b) spectra of a typical gold nanoparticle deposition. Spectra measured at position b from a bottom plate deposition of auric acid (0.12 mmol) in methanol (20 ml). | ||
The gold films possessed a broad reflectance from 500 nm and onwards towards longer wavelengths characteristic of the metallic reflectance. There is a strong absorbance from the SPR, resulting in the blue colour when looking through the film.
 | ||
| Fig. 6 Photograph showing the gradient deposition viewed from transmitted (top) and reflected light from an auric acid and butyltin trichloride ratio of 0.06 on the top plate. | ||
2.6 Results of Au:SnO2 composite films
Films were grown from a one pot solution of BuSnCl3 (2 mmol) and varying amounts of HAuCl4·3H2O dissolved in 20 ml of methanol as shown in Table 1. The Au:Sn ratio of 0.06 was then chosen for more in depth investigations at 400, 450 and 500 °C because the film had the best incorporation and gradient of gold in the SnO2 matrix (Fig. 6).
| MBTC amount/mmol | HAuCl4 amount/mmol | Au:Sn precursor atomic ratio |
Deposition temperature (°C) |
|---|---|---|---|
| 2 | 0 | 0 | 400, 450 and 500 |
| 0 | 0.12 | — | 450 |
| 2 | 0.1 | 0.05 | 500 |
| 2 | 0.12 | 0.06 | 400, 450 and 500 |
| 2 | 0.20 | 0.10 | 500 |
| 2 | 0.40 | 0.25 | 500 |
The bottom plate which rests on the carbon block is at the set temperature, the top plate however is typically around 50 °C lower in temperature. Film deposition occurred on the top plate, composed of mostly gold and the bottom plate, of mostly tin(IV) oxide. The films deposited at 400 °C and 450 °C tended to cover the whole substrate. At higher temperatures, i.e. 500 °C the precursor breaks down quicker and most of the deposition occurred in the first 2/3 of the substrate which relates to the regions investigated (Fig. 1).
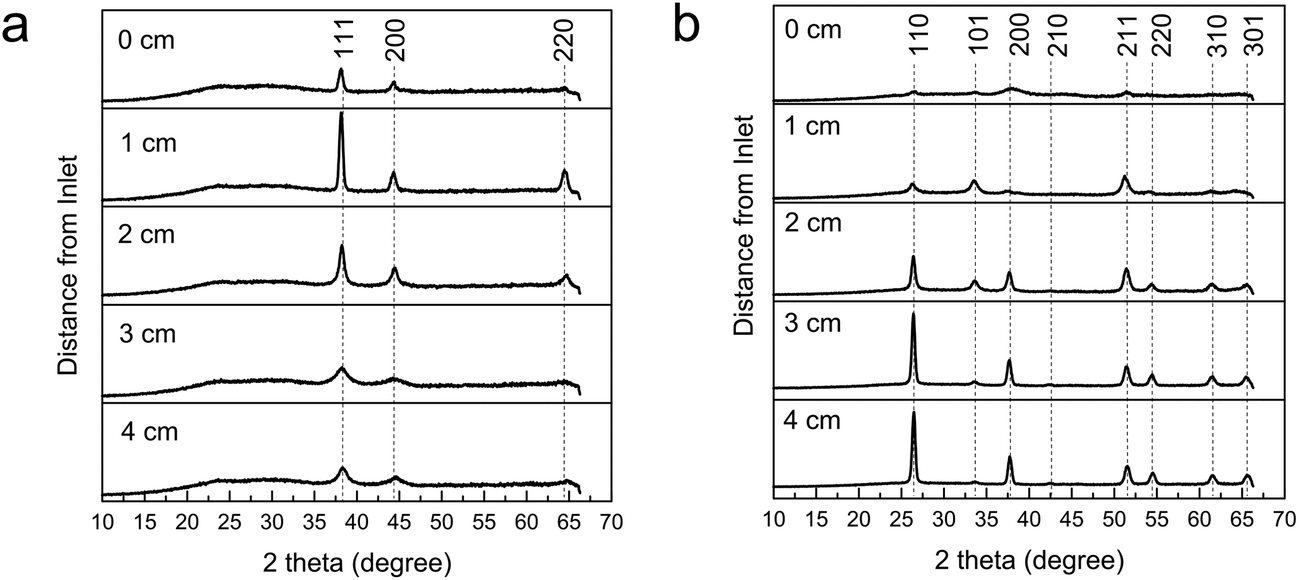 | ||
| Fig. 7 XRD patterns of the top-plate (a) and bottom-plate (b) of gold nanocomposite SnO2 (precursor 0.06 ratio) deposited at 450 °C. Labels refer to reflections from metallic Au (left figure) and SnO2 (right). | ||
Normally the presence of gold is not detectable in the XRD pattern of the bottom plate depositions even though gold is detected (EDX and XPS) and an SPR can be observed, thus the nanoparticles are believed to be embedded within the SnO2 matrix rather than on the surface. The broadening of the SnO2 reflections have been observed to be a result from (i) decreasing deposition temperature and so smaller crystallite size,26 and (ii) the addition of auric acid in the precursor solution which in the reactor, interferes with the surface reactions and deposition of crystalline SnO2. Overall, the top plates have lower amounts of deposit and the signal from the amorphous glass comes through in the XRD patterns resulting in broader peaks and higher backgrounds. Most of the XRD patterns of all the top plates display exclusively cubic Au even though the presence of SnO2 could be detected by XPS for the 500 °C deposition indicating a very thin film. If there was sufficiently high Au and SnO2 deposit, both of their peaks can be detected in XRD (see Fig. 8).
 | ||
| Fig. 8 XRD pattern showing a region of 0.5 Au:Sn (mol). Planes refer to SnO2 (*) and Au (○). | ||
EDX was used to quantify the Au:Sn atomic ratio of the deposited film on the heated substrate and is given in Table 1. XPS was also used to identify the elements. An example XPS for a deposition at 500 °C (region a) is shown in Fig. 9 where the calculated Au:Sn (mol) from the spectra is 0.52, agreeing well with EDX of 0.47. The Sn 3d peaks display an observed spin orbit coupling of 8.4 eV which is in good agreement with reference for SnO2.27 The binding energy of the 3d 5/2 is observed at 486.8 eV, which is comparable to the reported range of 486.95–487.3 eV.28–30 XPS also shows that the deposition of gold occurs on both the bottom and top plate and was dependant on location (see Table 2). The Au 4f 7/2 peak from the nanoparticles was located at 84.1 eV around the region of elemental gold reference values at 84.00 eV (ref. 31) and 84.04 eV.32 On the bottom plates, XPS gave no evidence for Sn incorporating in the gold nanoparticles or for gold substituting into the SnO2 lattice. The XPS data again confirmed the presence of mostly Au on the top plate as SnO2 was only detected for depositions at 500 °C.
 | ||
| Fig. 9 Photoelectron spectra from a bottom plate deposition (Au:Sn 0.06 precursor concentration at 500 °C) of Sn 3d 3/2 (495.9 eV) and 5/2 (487.5 eV) and Au 4f 5/2 (88.3 eV) and 4f 7/2 (84.6 eV) core levels. | ||
:Sn molar ratio which were determined by EDX. The corresponding SPR maxima were measured. All the films were deposited from a precursor solution of 2 mmol BuSnCl3 and a Au:Sn molar ratio of 0.06 in 20 ml methanol at 400–500 °C. The region labels (a–c) are stated with the deposition temperatures referred to in Fig. 1
| Temperature of deposition (°C) | Au:Sn (mol) |
SPR maximum (nm) | XRD phase observed | Lattice parameters of SnO2 unit cell a = b; c (Å) |
|---|---|---|---|---|
| 500 (a) | 0.47 | 559 | Au/SnO2 | 4.73; 3.21 |
| 500 (b) | 0.14 | 555 | SnO2 | 4.74; 3.18 |
| 500 (c) | 0.03 | — | SnO2 | 4.77; 3.19 |
| 450 (a) | 0.54 | 570 | Au | — |
| 450 (b) | 0.12 | 551 | Au/SnO2 | 4.77; 3.20 |
| 450 (c) | 0.08 | 558 | SnO2 | 4.74; 3.16 |
| 400 (a) | 0.50 | 597 | Au | — |
| 400 (b) | 0.21 | 517 | Au | — |
| 400 (c) | 0 | — | SnO2 | 4.78; 3.19 |
:Sn. The morphological changes from faceted to clusters were also observed for decreasing deposition temperature and increasing gold precursor. Higher temperatures seem to favour the formation of polycrystalline SnO2 on the bottom plate, which might be due to a larger amount of gold nanoparticles being thermophoretically pushed towards the top plate. For higher Au:Sn precursor ratio or at a lower temperature, there are more resulting gold nanoparticles in the reactor and on the bottom plate surface which might interrupt the SnO2 formation, resulting in clusters rather than strongly crystalline facets.
 | ||
| Fig. 10 SEM image taken at different regions on the bottom plate of Au-doped SnO2 deposited at 500 °C from a precursor solution of 0.06 Au:Sn atomic ratio in 20 ml methanol – distance from inlet (a) 0 cm, (b) 4 cm and (c) 8 cm. | ||
Individual gold particles were observed on the top plate of films deposited at 400 and 450 °C and a layer of nanoparticles for films deposited at 500 °C (see Fig. 11). As mentioned before, thermophoresis has an increase effect on larger particles, it indicates that the gold atoms are aggregating and forming nanoparticles in gas phase reactions before deposition. As with XRD patterns, the gold nanoparticles cannot be seen in the SEM images of the bottom plates even when an SPR absorption was present, indicating they are embedded within the SnO2 host matrix.
 | ||
| Fig. 11 SEM images of gold nanoparticles deposited on the top plate with HAuCl4·3H2O and BuSnCl3 ratio of 0.06 in 20 ml methanol at (a) 500 °C, (b) 450 °C, and (c) 400 °C. | ||
 | ||
| Fig. 12 Transmission (i), absorption (ii) and reflection (iii) spectra corresponding to depositions from a precursor solution of 0.06 Au:Sn atomic ratio at 500 °C in 20 ml methanol on the bottom plate. The ratio of Au:Sn and the regions investigated as a distance from the inlet (a) 0 cm, (b) 4 cm and (c) 8 cm are labelled. | ||
Fig. 12(i) shows the transmission from the bottom plate deposited at 500 °C from a Au:Sn atomic ratio of 0.06 in the precursor solution at distances of around 1, 5 and 9 cm away from the inlet chosen for their measured Au:Sn (EDX) and corresponding to the data in Table 2. The graph in Fig. 12(ii) shows the corresponding SPR band absorptions. Higher Au:Sn ratio results in a lower transmission across all recorded wavelengths and higher relative absorbance intensities.
UV/visible spectroscopy showed surface plasmon (SPR) absorption peaks in all the films but not all areas on the bottom and top plates. There were areas where the interference patterns were strong and the SPR peaks could not be determined. The width of the SPR band increase with Au content indicating a widening of the dispersion of the nanoparticles.33 From the observable SPR bands, the maxima varied and ranged from 510 to 602 nm also indicating a variety of sizes and concentrations. The position of the SPR usually changes according to the different sizes of the nanoparticles, the concentration and the dielectric constant of the medium in which they sit in. However, due to the fact that the nanoparticles in the Au/SnO2 are embedded within the metal oxide matrix, the sizes and concentration of the nanoparticles could not be investigated and correlated with the SPR position.
On the top plate where only gold depositions were seen, there is an opposite trend (ESI Fig. 1†). As EDX shows no Sn deposited, no Au:Sn value could be measured and the absorbance intensity was taken as an indication of the amount of gold deposited. An increase in gold content thus results in a narrowing of the spectroscopic SPR, this narrowing suggests lower dispersity.33 This is similar to what is seen with depositions of gold films, where a lower precursor concentration has been shown to give island growth morphology whilst a higher concentration shows particulate morphology,24 due to gas phase reactions being favoured at high concentrations and surface reactions at lower concentrations.
The methanol carrier solvent has a key effect in the reaction – both for formation of the nanoparticles and for the metal oxide host formation. Methanol has been shown in a number of CVD reactions to promote the formation of elemental metals – especially for Cu,34 Ag (ref. 35) and Au.24 It is thought that it can decompose in the reactor in the presence of metal nanoparticles to form an in situ source of hydrogen – which may aid the formation of the nanoparticles. Conversely the methanol also acts as the source of oxygen for the formation of the tin oxide matrix as the reaction was conducted under a nitrogen atmosphere. One possible set of equations for the processes are schematically illustrated below;
| SnBuCl3 + 4MeOH → SnO2 + 3HCl + 2Me–O–Me + Bu–H |
| 2HAuCl4 + 6MeOH → 2Au + 8HCl + 3Me–O–Me + 1.5 O2 |
If using this set up and only the bottom plate was used there would be a waste in material leading this to be classified as an inefficient method therefore both top and bottom plates were analysed. We see an increase in dispersity of the top plate nanoparticles when starting with a low Au precursor concentration. This is in contrast to bottom plate nanoparticles where the dispersity decreases with gold content due to stabilisation of SnO2 and the decrease in mobility of Au particles. The presence of the SPR and the elemental detection of gold (EDX/XPS) suggests the presence of gold nanoparticles despite not being able to readily image them in SEM images. The relationship between the SPR width and the Au content promises an ability to tune the characteristics of the nanoparticles with deposition conditions.
3 Conclusion
Composite Au/SnO2 films were grown from a simple one pot precursor solution using aerosol assisted CVD. Two films were formed, mostly gold films on the top plate and Au/SnO2 films on the heated substrate. The composite film showed different amounts of gold incorporation with position from the inlet affording us investigations into a graded film which enabled us rapid assessment of the films functional properties at different deposited Au:Sn. These films with a Au:Sn gradation could be reproduced when using identical experimental procedures. Optimum locations were found at regions with 0.03–0.15 Au:Sn molar ratio, where a combination of desirable properties were shown including a blue colouration, reasonable optical transparency as well as reflectivity in the IR, ideal for a tinted low emissivity window coating. Such films could find application in commercial float glass production. The tint of the film was dependant on the concentration of nanoparticles incorporated and is also affected by the dielectric constant of the SnO2 matrix which could be explored further. One issue that will need to be overcome however is the relative degree of incorporation of gold into the film versus the amount lost to exhaust. The fact that a gold film could be formed on a top plate and potentially recovered provides a better material cost balance, especially as the gold nanoparticles can be relatively easily removed. Fluorine doping of SnO2 will improve the conductivity, but because the nanoparticles are embedded in the SnO2 creating resistance effects, a layered thin film approach could eliminate this.
References
- U. Kreibig and M. Vollmer, Optical Properties of Metal Clusters, Springer, 1995 Search PubMed.
- S. Cho, S. Lee, S. Oh, S. J. Park, W. M. Kim, B. Cheong, M. Chung, K. B. Song, T. S. Lee and S. G. Kim, Thin Solid Films, 2000, 377–378, 97–102 CrossRef CAS.
- N. Félidj, J. Aubard, G. Lévi, J. R. Krenn, A. Hohenau, G. Schider, A. Leitner and F. R. Aussenegg, Appl. Phys. Lett., 2003, 82, 3095 CrossRef PubMed.
- C. Gomes Silva, R. Juárez, T. Marino, R. Molinari and H. García, J. Am. Chem. Soc., 2011, 133, 595–602 CrossRef CAS PubMed.
- J. Deng, M. Gu and J. Di, Appl. Surf. Sci., 2011, 257, 5903–5907 CrossRef CAS PubMed.
- P. Manjula, S. Arunkumar and S. V. Manorama, Sens. Actuators, B, 2011, 152, 168–175 CrossRef CAS PubMed.
- G. Walters and I. P. Parkin, J. Mater. Chem., 2009, 19, 574 RSC.
- C. M. Lampert, Sol. Energy Mater. Sol. Cells, 2003, 76, 489–499 CrossRef CAS.
- K. Ellmer, J. Phys. D: Appl. Phys., 2001, 34, 3097–3108 CrossRef CAS.
- C. G. Granqvist and A. Hultåker, Thin Solid Films, 2002, 411, 1–5 CrossRef CAS.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2012, 99, 1–13 CrossRef CAS PubMed.
- T. Oyabu, J. Appl. Phys., 1982, 53, 7125 CrossRef CAS PubMed.
- T. T. Stoycheva, S. Vallejos, R. G. Pavelko, V. S. Popov, V. G. Sevastyanov and X. Correig, Chem. Vap. Deposition, 2011, 17, 247–252 CrossRef CAS.
- T. Maruyama and K. Tabata, J. Appl. Phys., 1990, 68, 4282 CrossRef CAS PubMed.
- J. C. Manifacier, L. Szepessy, J. F. Bresse, M. Perotin and R. Stuck, Mater. Res. Bull., 1979, 14, 163–175 CrossRef CAS.
- NanoMarkets, Transparent Conductor Markets 2010: ITO and the Alternatives, 2010 Search PubMed.
- T. Kawashima, T. Ezure, K. Okada, H. Matsui, K. Goto and N. Tanabe, J. Photochem. Photobiol., A, 2004, 164, 199–202 CrossRef CAS PubMed.
- C. G. Granqvist, Sol. Energy Mater. Sol. Cells, 2007, 91, 1529–1598 CrossRef CAS PubMed.
- P. Marchand, I. A. Hassan, I. P. Parkin and C. J. Carmalt, Dalton Trans., 2013, 42, 9406–9422 RSC.
- K. C. Molloy, J. Chem. Res., 2008, 549–554 CrossRef CAS.
- Jacques Dela Ruye, US Pat., 4339541, 1982.
- R. Binions, C. Piccirillo, R. G. Palgrave and I. P. Parkin, Chem. Vap. Deposition, 2008, 14, 33–39 CrossRef CAS.
- T. Ung, L. M. Liz-Marzán and P. Mulvaney, Colloids Surf., A, 2002, 202, 119–126 CrossRef CAS.
- R. G. Palgrave and I. P. Parkin, Chem. Mater., 2007, 19, 4639–4647 CrossRef CAS.
- Y. Matsui, M. Mitsuhashi, Y. Yamamoto and S. Higashi, Thin Solid Films, 2007, 515, 2854–2859 CrossRef CAS PubMed.
- N. Noor and I. P. Parkin, J. Mater. Chem. C, 2013, 1, 984 RSC.
- J. C. Fuggle and N. Mårtensson, J. Electron Spectrosc. Relat. Phenom., 1980, 21, 275–281 CrossRef CAS.
- W.-K. Choi, J. Vac. Sci. Technol., A, 1996, 14, 359 CAS.
- S. Badrinarayanan, A. B. Mandale, V. G. Gunjikar and A. P. B. Sinha, J. Mater. Sci., 1986, 21, 3333–3338 CrossRef CAS.
- S. Süzer, T. Voscoboinikov, K. R. Hallam and G. C. Allen, Anal. Bioanal. Chem., 1996, 355, 654–656 CrossRef PubMed.
- M. P. Seah, G. C. Smith and M. T. Anthony, Surf. Interface Anal., 1990, 15, 293–308 CrossRef CAS.
- T. Thomas and P. Weightman, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 5405–5413 Search PubMed.
- M. A. Uppal, A. Kafizas, T. H. Lim and I. P. Parkin, New J. Chem., 2010, 34, 1401 RSC.
- C. R. Crick and I. P. Parkin, J. Mater. Chem., 2011, 21, 14712 RSC.
- A. Panneerselvam, M. A. Malik, P. O'Brien and J. Raftery, J. Mater. Chem., 2008, 18, 3264 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra46828c |
| This journal is © The Royal Society of Chemistry 2014 |