
Synthesis and photocatalytic activity of a bentonite/g-C3N4 composite
Abstract
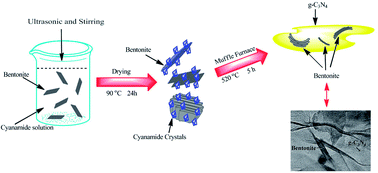
Novel bentonite/g-C3N4 composite photocatalysts were synthesized through a conventional calcination method and systematically characterized by thermogravimetric analysis (TG), powder X-ray diffraction (XRD), transmission electron microscopy (TEM), Fourier transform infrared spectroscopy (FT-IR), UV-vis diffuse reflection spectroscopy (DRS), X-ray photoelectron spectroscopy (XPS), photoluminescence (PL) and photocurrent–time measurement (PT). The optimal sample bentonite/g-C3N4 (5.4%) composite shows an enhanced photocurrent value (about 8 times as high as that of g-C3N4) under visible light irradiation and high efficiency for the degradation of methylene blue (the photoreaction kinetics constant value is about 2.5 times that of g-C3N4) under visible light. Results show the improved photoactivity is mainly attributed to the electrostatic interaction between g-C3N4 and negatively charged bentonite, this leads to the efficient migration of the photogenerated electrons and holes of g-C3N4. This study reports an inexpensive and environmentally friendly photocatalyst for pollution degradation and prospective photoelectric materials.
 Please wait while we load your content...
Please wait while we load your content...

