1-Aryl-1,2,3,4-tetrahydroisoquinolines as potential antimalarials: synthesis, in vitro antiplasmodial activity and in silico pharmacokinetics evaluation†
Joelle Ngo Hanna‡
a,
Fidele Ntie-Kanga,
Marcel Kaiserb,
Reto Brunb and
Simon M. N. Efange*a
aDepartment of Chemistry, Faculty of Science, University of Buea, P.O. Box 63, Buea, South West Region, Cameroon. E-mail: smbuangalefange@gmail.com; Tel: +237 99915610
bDepartment of Medical Parasitology and Infection Biology Parasite Chemotherapy, Swiss Tropical Institute, Basel, Switzerland
First published on 3rd April 2014
Abstract
In the present study, twenty-one 1-aryl-6-hydroxy-1,2,3,4-tetrahydroisoquinoline (THIQ) analogues were synthesized by base-catalyzed Pictet–Spengler reaction, and tested in vitro against P. falciparum using the [3H]hypoxanthine incorporation assay. Two compounds were found to be inactive while seventeen compounds displayed moderate antiplasmodial activity and two compounds were found to be highly active (IC50 < 0.2 μg ml−1). The two highly active compounds, 1-(4-chlorophenyl)-6-hydroxyl-1,2,3,4-tetrahydroisoquinoline and 6-hydroxyspiro[1,2,3,4-tetrahydroisoquinoline-1:1′-cyclohexane], also displayed low cytotoxicity, against rat skeletal myoblast cells, with CC50 values of 257.6 and 174.2 μM respectively. These results justify further investigation of simple 1-aryl-1,2,3,4-tetrahydroisoquinolines as potential anti-malarial agents.
Introduction
Malaria is a life threatening infectious parasitic disease that inflicts both health and economic losses.1 The disease is transmitted by mosquitoes of the genus Anopheles. The causative agent for malaria is the Plasmodium species, and there are four types of human malaria: P. vivax: P. malaria, P. ovale and P. falciparum. P. vivax and P. falciparum are the most common, and P. falciparum is responsible for the most deadly type of malaria infection.2 Statistics indicate that P. falciparum causes more than 300 to 500 million clinical cases of malaria infection, resulting in 1.7 to 2.5 million deaths annually.3 Malaria is a particularly important disease in Sub-Saharan Africa where about 90% of cases and death occur, but is also a serious public health burden in South, East and Central Asia, South and Central America, Caribbean, Oceania, and Middle East regions.4 Many attempts have been made to control the disease by using vector control measures and/or chemoprophylaxis, but they have had limited success.5 The control of malaria is, among other factors, hampered by the emergence of resistance of the vector (Anopheles sp.) to dichlorodiphenyltrichloroethane (DDT) and other insecticides, and the ongoing spread of strains of P. falciparum that are resistant to currently used anti-malarial drugs.6 Attempts to address the problem of resistance have resulted in the current shift to artemisinin based combination drug regimens. New anti-malarial drugs, especially those with new structures and novel modes of action against malaria are also needed. This work describes the synthesis of structurally simple tetrahydroisoquinolines (THIQs) and their preliminary evaluation as potential anti-malarial agents. THIQs abound in nature and have attracted significant attention due to their diverse biological activities. The THIQ substructure (Fig. 1) is found in many drugs and alkaloids that exhibit antitumor, cardiovascular, antibacterial, antimicrobial, antitubercular, antifungal, antileishmanial, antitrypanosomal and antiplasmodial activities.7–20 These compounds include the naphthylisoquinolines, benzylisoquinolines and bisbenzylisoquinolines. | ||
| Fig. 1 1,2,3,4-Tetrahydroisoquinoline. | ||
The broad spectrum of biological activities of tetrahydroisoquinolines, especially reports on the anti-human immunodeficiency virus (HIV) activity of michellamine B (1) and antiplasmodial activity of dioncophylline C (3), naphthylisoquinolines (Fig. 2) isolated from Ancistrocladus korupensis,11 prompted us to investigate 1-aryl-1,2,3,4-tetrahydroisoquinolines, as abbreviated analogues of the naturally occurring compounds, for antiplasmodial activity. Naphthylisoquinoline alkaloids are known to exhibit curative potentials against malaria.12–14 Dioncophylline E (5) has exhibited good antimalarial activity against both chloroquine-sensitive and -resistant strains of P. falciparum in vitro,13 while dioncophylline C was active against the chloroquine-resistant P. berghei Anka CRS parasites12 with dioncophylline B (2), dioncophylline D (4) and synthetic naphthylisoquinolines also found to display good in vitro antiplasmodial activities.14 A noteworthy example of a structurally simple THIQ is the spirofused compound IVa, which was previously described by Kametani et al.,21 as an abbreviated analogue of erysodine, one member of the Erythrina class of alkaloids. Plants of the genus Erythrina have been used extensively in ethnomedicine for a number of applications including the treatment of liver disorders,20 as antiplasmodial agents, analgesics, and anti-inflammatory agents,21 as well as in the treatment of diseases such as female infertility, stomach pain and gonorrhea.22 Moreover, the alkaloids of Erythrina type are characterized by their unique skeleton of a tetracyclic spiroamine.23 They are classified in two main groups: alkaloids possessing a skeleton of a 6,5,6,6 indoloisoquinoline called erythrinanes and those with a skeleton of 6,5,7-6 indolobenzazepine called schelhammerans or homoerythrinane alkaloids (Fig. 3). Erythrina alkaloids are known to display antiplasmodial activity in vitro.24–26 The rationale behind the design of THIQ analogues as potential anti-malarials is that they seem to be the common denominator of the naturally occurring naphthoisoquinolines (1 to 5) which display anti-malarial activities (marked in red on Fig. 2). It could therefore be easily inferred that the THIQ scaffold could contain the pharmacophore responsible for the anti-malarial activity. Previous studies have revealed that even simple THIQs such as 1-aryl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinolines and 1-aryl-6,7-dimethoxy-1,2,3,4-tetrahydroisquinolines, display anti-HIV15 and bronchodilator16 properties. The current study, which forms part of a larger investigation, was undertaken to investigate the antiplasmodial activity of 1-aryl-1,2,3,4-tetrahydroisoquinolines. The THIQ skeleton presents eight positions that can accommodate substituents. The choice of positions 1 and 6 for this study was based on the ease of synthesis of 1-substituted-THIQs (which is offered by the Pictet Spengler) and the presence of alkoxy groups on the aromatic portion of complex THIQs like the naphthoisoquinolines, such as 2–5, which display antiplasmodial activity.
 | ||
| Fig. 2 Michellamine B (1) and dioncophyllines B (2), C(3), D(4) and E(5) with the tetrahydroisoquinoline scaffold shown in red. | ||
 | ||
| Fig. 3 Homoerythrinane alkaloids. | ||
Designing structurally simple THIQs would therefore considerably lower the molecular masses and hence produce analogues with properties that fall within the range of those of ‘drug-like’ compounds. The target 6-hydroxy-1,2,3,4-tetrahydroisoquinoline derivatives (IIIa to IIIu and IVa) were synthesized via base-catalyzed Pictet–Spengler cyclization as previously reported.19 Overall yields over two steps ranged between 4 and 99%. The compounds were characterized by NMR and mass spectrometry. The corresponding hydrochlorides were subsequently tested in vitro for antiplasmodial activity against P. falciparum. An in silico assessment of drug metabolism and pharmacokinetics (DMPK) profiles was also carried out by the use of computed molecular descriptors related to the absorption, distribution, metabolism, excretion and toxicity (ADMET) of compounds.
Results and discussion
Antiplasmodial activity versus cytotoxicity
All compounds were tested in vitro against P. falciparum as described in the Experimental section. The biological data were plotted to obtain IC50 values which represent the concentration required to cause 50% inhibition of parasite growth. Activity criteria were as follows: inactive, IC50 > 5 μg mL−1; moderately active, 0.5 μg mL−1 < IC50 < 5 μg mL−1; highly active, IC50 < 0.5 μg mL−1. The antiplasmodial activity, presented as IC50 values, is provided in Table 1. The P. falciparum strain K1, known to be resistant to chloroquine and pyrimethamine, was employed for this assay. Chloroquine was used as the reference drug.| Compounds | Substituent (R) | IC50 P. falciparum K1 (μg mL−1) | IC50 P. falciparum K1 (μM) | Cytotoxicity L6 (μg mL−1) | Cytotoxicity L6 (μM) | Selectivity index |
|---|---|---|---|---|---|---|
| a NA: not available. | ||||||
| IIIa | H | 0.519 | 2.304 | >90 | >400 | >200 |
| IIIb | 4-Cl | 0.181 | 0.697 | 66.9 | 257.6 | 369.6 |
| IIIc | 3-Cl | 0.347 | 1.336 | 48.9 | 188.3 | 140.9 |
| IIId | 3,4-Cl2 | 0.395 | 1.343 | 29.1 | 98.9 | 73.7 |
| IIIe | 3-NO2 | 0.347 | 1.284 | >90 | >400 | >300 |
| IIIf | 3-OCH3 | 0.646 | 2.530 | 84.9 | 332.5 | 131.4 |
| IIIg | 2,3-(OCH3)2 | 0.892 | 3.126 | >90 | >400 | >100 |
| IIIh | 2,5-(OCH3)2 | 1.22 | 4.276 | >90 | >400 | >100 |
| IIIi | 2,6-(OCH3)2 | >5 | >20 | >90 | >400 | >200 |
| IVa | Spiro | 0.193 | 0.885 | >90 | >400 | >400 |
| IIIj | 4-Br | 0.481 | 1.581 | 47.44 | 155.9 | 98.6 |
| IIIk | 4-CF3 | 0.223 | 0.760 | 51.1 | 174.2 | 229.1 |
| IIIl | 4-Phenyl | 0.906 | 3.006 | 3.29 | 10.9 | 3.6 |
| IIIm | 2-Br | NA | NA | NA | NA | NA |
| IIIn | 2-F | 2.31 | 9.495 | 28.3 | 116.3 | 12.3 |
| IIIo | 3-Br | 1.08 | 3.550 | 9.06 | 29.8 | 8.4 |
| IIIp | 3-F | 1.59 | 6.787 | 33.1 | 141.3 | 20.8 |
| IIIq | 4-F | 0.595 | 2.446 | 36.5 | 150.0 | 61.3 |
| IIIr | 4-Cl-3-NO2 | 2.05 | 6.727 | 24.9 | 81.7 | 12.1 |
| IIIs | 5-Br-2-OCH3 | 0.744 | 2.226 | 9.18 | 27.5 | 12.3 |
| IIIt | 2-OH-5-NO2 | 10.3 | 35.978 | 19.5 | 68.1 | 1.9 |
| IIIu | 4-CH3 | 0.891 | 3.955 | 21.6 | 95.9 | 24.2 |
| Chloroquine | 0.061 | 0.192 | NA | NA | 3000 | |
| Podophyllotoxin | 0.004 | |||||
Of twenty-one compounds tested, three could be described as inactive following the criteria established. The rest were either moderately active (twelve) or active (seven). Among the active compounds at least three (IIIb, IIIk and IVa) displayed antiplasmodial activity comparable to that of chloroquine, suggesting the need for further evaluation of these compounds in vivo. In general, the more active compounds tended to possess electron withdrawing substituents on the pendant phenyl group (IIIb-e,j,k,q). However, a number of moderately active compounds contained electron donating substituents on the same phenyl group, thereby obscuring the role of electronic substituent parameters as determinants of antiplasmodial activity in this series. The present demonstration of antiplasmodial activity in the compound would therefore suggest that the key pharmacophoric elements for antiplasmodial activity in this class of alkaloids reside in the skeleton as described by IVa.19
When tested for cytotoxicity, several of the compounds were found to be considerably less toxic against rat skeletal mycoblast cells than the reference compound podophyllotoxin. In addition, the tested compounds displayed toxicity at concentrations that were two orders of magnitude higher than the concentrations at which they displayed antiplasmodial activity. Therefore, structurally simplified 1,2,3,4-tetrahydroisoquinolines may be potentially useful as antiplasmodial agents.
Compounds IIIb, IIIk and IVa were the most active with IC50 values 0.70, 0.76 and 0.89 μM, respectively, and their activity was in the same range as that of chloroquine. In addition, the cytotoxicity of these compounds against myoblast cells could be considered as an index of drug effectiveness. Compounds IIIb, IIIk and IVa had selectivity index values 369.6, 229.1 and >451.5 respectively. These values are up to tenfold lower than the selectivity index value reported for chloroquine but significantly higher than the value of 100 which is required to qualify a compound as a “hit” in the screening of compounds for antiplasmodial activity.
At 2.30 μM, IIIa was moderately active, but the presence of chlorine at C-4 resulted in a significant increase of activity, maybe due to the electron withdrawing property by inductive effect and ortho and para directive effect. This activity is not maintained in IIIc, IIId, IIIe, IIIj, IIIm, IIIn, IIIo, IIIp and IIIq. When a methoxyl group (electron donating by resonance) is introduced in compound IIIa to get IIIf, only a slight drop in activity is observed. When additional methoxyl groups are introduced, (IIIg, IIIh, IIIi) the activity drops even further. In general, the effects of both mono- and di-substitution on the pendant phenyl group appeared to be largely dependent on the nature of the substituent(s), but C4-substitution, especially with electron withdrawing groups of moderate size, appeared to be the most favored substitution pattern (compare IIIq vs IIIp vs IIIn; IIIb vs IIIc and IIIj vs IIIo). The favorable antiplasmodial activity of spirocylohexyl analogue IVa may be attributed to the increased rigidity of this analogue.
In silico pharmacokinetics profiles
Molecular modelling was carried out on all synthesised molecules as described in the Experimental section. The computed molecular descriptors are those related to the prediction of DMPK/ADMET properties like the n-octanol–water partition coefficient, aqueous solubility, brain–blood partition coefficient, Caco-2 cell permeability, serum protein binding, number of likely metabolic reactions, solvent accessible surface area, blockage of human-ether-a-go-go potassium ion (HERG K+) channels and the total volume of the molecule enclosed by the solvent accessible surface area. The bioavailability of a compound depends on the processes of absorption and liver first-pass metabolism.27 Absorption in turn depends on the solubility and permeability of the compound, as well as interactions with transporters and metabolizing enzymes in the gut wall. The computed parameters used to assess oral absorption are the predicted aqueous solubility, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Swat, the conformation-independent predicted aqueous solubility, CIlogSwat, the predicted qualitative human oral absorption, the predicted % human oral absorption and compliance to Lipinski's famous “Rule of Five” (ro5).28 The size of a molecule, as well as its capacity to make hydrogen bonds, its overall lipophilicity and its shape and flexibility are important properties to consider when determining permeability. Molecular flexibility has been seen as a parameter which is dependent on the number of rotatable bonds (NRB), a property which influences bioavailability in rats.29 The blood–brain partition coefficients (logB/B) were computed and used as a predictor for access to the central nervous system (CNS). Madin–Darby canine kidney (MDCK) monolayers, are widely used to make oral absorption estimates, the reason being that these cells also express transporter proteins, but only express very low levels of metabolizing enzymes.29 They are also used as an additional criterion to predict BBB penetration. Thus, our calculated apparent MDCK cell permeability could be considered to be a good mimic for the BBB (for non-active transport). The efficiency of a drug may be affected by the degree to which it binds to the proteins within blood plasma. Binding of drugs to plasma proteins (like human serum albumin, lipoprotein, glycoprotein, α, β‚ and γ globulins) greatly reduces the quantity of the drug in general blood circulation and hence the less bound a drug is, the more efficiently it can traverse cell membranes or diffuse. The predicted plasma-protein binding has been estimated by the prediction of binding to human serum albumin; the logKHSA parameter (recommended range is −1.5 to 1.5 for 95% of known drugs). Human ether-a-go-go related gene (HERG) encodes a potassium ion (K+) channel that is implicated in the fatal arrhythmia known as torsade de pointes or the long QT syndrome.30 The HERG K+ channel, which is best known for its contribution to the electrical activity of the heart that coordinates the heart's beating, appears to be the molecular target responsible for the cardiac toxicity of a wide range of therapeutic drugs.32 HERG has also been associated with modulating the functions of some cells of the nervous system and with establishing and maintaining cancer-like features in leukemic cells.32 Thus, HERG K+ channel blockers are potentially toxic and the predicted IC50 values often provide reasonable predictions for cardiac toxicity of drugs in the early stages of drug discovery.33
Swat, the conformation-independent predicted aqueous solubility, CIlogSwat, the predicted qualitative human oral absorption, the predicted % human oral absorption and compliance to Lipinski's famous “Rule of Five” (ro5).28 The size of a molecule, as well as its capacity to make hydrogen bonds, its overall lipophilicity and its shape and flexibility are important properties to consider when determining permeability. Molecular flexibility has been seen as a parameter which is dependent on the number of rotatable bonds (NRB), a property which influences bioavailability in rats.29 The blood–brain partition coefficients (logB/B) were computed and used as a predictor for access to the central nervous system (CNS). Madin–Darby canine kidney (MDCK) monolayers, are widely used to make oral absorption estimates, the reason being that these cells also express transporter proteins, but only express very low levels of metabolizing enzymes.29 They are also used as an additional criterion to predict BBB penetration. Thus, our calculated apparent MDCK cell permeability could be considered to be a good mimic for the BBB (for non-active transport). The efficiency of a drug may be affected by the degree to which it binds to the proteins within blood plasma. Binding of drugs to plasma proteins (like human serum albumin, lipoprotein, glycoprotein, α, β‚ and γ globulins) greatly reduces the quantity of the drug in general blood circulation and hence the less bound a drug is, the more efficiently it can traverse cell membranes or diffuse. The predicted plasma-protein binding has been estimated by the prediction of binding to human serum albumin; the logKHSA parameter (recommended range is −1.5 to 1.5 for 95% of known drugs). Human ether-a-go-go related gene (HERG) encodes a potassium ion (K+) channel that is implicated in the fatal arrhythmia known as torsade de pointes or the long QT syndrome.30 The HERG K+ channel, which is best known for its contribution to the electrical activity of the heart that coordinates the heart's beating, appears to be the molecular target responsible for the cardiac toxicity of a wide range of therapeutic drugs.32 HERG has also been associated with modulating the functions of some cells of the nervous system and with establishing and maintaining cancer-like features in leukemic cells.32 Thus, HERG K+ channel blockers are potentially toxic and the predicted IC50 values often provide reasonable predictions for cardiac toxicity of drugs in the early stages of drug discovery.33
The results for fifteen (15) relevant descriptors have been shown in Table 2, while 2D scatter plots of selected molecular descriptors are shown in Fig. 4. It was observed that only the points of subfigure C fell outside the recommended range for the dipole moment parameter. Subfigures A and B showed that all the compounds complied with Lipinski's ro5, since the molecular weights (MW) < 500 Da, logarithm of n-octanol–water partition coefficient (logP) < 5, number of hydrogen bond acceptors (accHB) < 10 and number of hydrogen bond donors (donHB) < 5. It was observed that the other computed molecular descriptors of all the THIQ analogues fell within the recommended range for 95% of known drugs (recommended parameters are shown in the ESI†), except for two compounds (IIIo and IIIq), as can be seen in subfigures 4D-L, corresponding respectively to the scatter plots for MW against the number of rotatable bonds (#rotor), cohension index in solids, blood–brain barrier partitioning, HERG K+ ion channel blockage, binding affinities to human serum albumin, skin permeability, aqueous solubilities, as well Caco-2 and MDCK permeabilities. As for compound IIIo, the dipole moment was computed to be 0.524 Debye, below the recommended range (1.0 to 12.5 Debye), while for compound IIIq, the computed value was 0.890 Debye, only slightly below the recommended minimum for 95% of known drugs. The weak dipole moments could be explained by the absence of polar groups in the pendant phenyl ring of the two aforementioned compounds. Dipole moments as high as >6 Debye were observed for compounds IIIe, IIIr and IIIt containing the polarising nitro group in the pendant phenyl ring. It is however important to mention that the most active compounds (IIIb, IIIk and IVa) had interesting predicted pharmacokinetic profiles (with all computed parameters falling within the recommended range for 95% of known drugs, i.e., #stars = 0). We can therefore conclude that these three compounds could be subjected to further investigation as potential anti-malarial drug leads.
| Compound | #starsb | Ro5c | MW (Da)d | logPe |
HBAf | HBDg | NRBh | logB/Bi |
BIPcaco-2j (nm s−1) | Smolk (Å2) | Smol,hfobl (Å2) | Vmolm (Å3) | logSwatn |
logKHSAo |
logHERGp |
#metabq |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a Property which falls outside the recommended range for 95% of known drugs.b Number of computed properties which fall outside the required range for 95% of known drugs.c Number of violations of Lipinski's “Rule of Five”.d Molar weight (range for 95% of drugs: 130–725 Da).e Logarithm of partitioning coefficient between n-octanol and water phases (range for 95% of drugs: −2 to 6.5).f Number of hydrogen bonds accepted by the molecule (range for 95% of drugs: 2–20).g Number of hydrogen bonds donated by the molecule (range for 95% of drugs: 0–6).h Number of rotatable bonds (range for 95% of drugs: 0–15).i Logarithm of predicted blood–brain barrier partition coefficient (range for 95% of drugs: −3.0 to 1.0).j Predicted apparent Caco-2 cell membrane permeability in Boehringer–Ingelheim scale, in nm s−1 (range for 95% of drugs: <5 low, >500 high).k Total solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 300–1000 Å2).l Hydrophobic portion of the solvent-accessible molecular surface, in Å2 (probe radius 1.4 Å) (range for 95% of drugs: 0–750 Å2).m Total volume of molecule enclosed by solvent-accessible molecular surface, in Å3 (probe radius 1.4 Å) (range for 95% of drugs: 500–2000 Å3).n Logarithm of aqueous solubility (recommended range is −6.0 to 0.5).o Logarithm of predicted binding constant to human serum albumin (range for 95% of drugs: −1.5 to 1.5).p Predicted IC50 value for blockage of HERG K+ channels (concern < −5).q Number of likely metabolic reactions (range for 95% of drugs: 1–8). | ||||||||||||||||
| IIIa | 0 | 0 | 225.29 | 2.37 | 2 | 2 | 1 | 0.27 | 496.43 | 471.63 | 108.05 | 791.71 | −2.47 | 0.20 | −5.62 | 4 |
| IIIb | 0 | 0 | 259.73 | 2.88 | 2 | 2 | 1 | 0.43 | 490.96 | 490.97 | 109.67 | 831.60 | −3.11 | 0.30 | −5.47 | 4 |
| IIIc | 0 | 0 | 259.73 | 3.04 | 2 | 2 | 1 | 0.43 | 502.08 | 496.04 | 110.47 | 834.22 | −3.21 | 0.30 | −5.61 | 3 |
| IIId | 0 | 0 | 294.18 | 3.35 | 2 | 2 | 1 | 0.56 | 491.91 | 512.32 | 109.83 | 875.21 | −3.74 | 0.41 | −5.42 | 3 |
| IIIe | 0 | 0 | 270.29 | 1.69 | 3 | 2 | 2 | −0.67 | 61.31 | 506.66 | 110.05 | 859.53 | −2.68 | 0.18 | −5.56 | 5 |
| IIIf | 0 | 0 | 255.32 | 2.52 | 3 | 2 | 2 | 0.21 | 493.67 | 501.58 | 201.08 | 870.15 | −2.65 | 0.25 | −5.30 | 5 |
| IIIg | 0 | 0 | 285.34 | 2.71 | 4 | 2 | 3 | 0.17 | 560.95 | 539.25 | 271.61 | 944.75 | −2.94 | 0.29 | −5.42 | 6 |
| IIIh | 0 | 0 | 285.34 | 2.63 | 4 | 2 | 3 | 0.11 | 496.58 | 543.12 | 292.28 | 943.14 | −3.01 | 0.28 | −5.42 | 6 |
| IIIi | 0 | 0 | 285.34 | 2.63 | 4 | 2 | 3 | 0.11 | 498.58 | 547.12 | 298.28 | 951.14 | −2.94 | 0.28 | −5.42 | 6 |
| IVa | 0 | 0 | 218.15 | 2.62 | 1 | 2 | 1 | 0.15 | 458.68 | 537.76 | 215.53 | 925.08 | −2.65 | 0.27 | −5.13 | 5 |
| IIIj | 0 | 0 | 304.19 | 2.89 | 2 | 2 | 1 | 0.44 | 491.10 | 494.37 | 109.56 | 839.44 | −3.02 | 0.32 | −5.47 | 4 |
| IIIk | 0 | 0 | 293.29 | 3.36 | 2 | 2 | 1 | 0.53 | 493.13 | 520.84 | 110.25 | 889.70 | −3.85 | 0.45 | −5.58 | 3 |
| IIIl | 0 | 0 | 301.39 | 3.95 | 2 | 2 | 2 | 0.13 | 495.30 | 589.39 | 109.78 | 1022.46 | −4.39 | 0.76 | −6.90 | 3 |
| IIIm | 0 | 0 | 304.19 | 2.89 | 2 | 2 | 1 | 0.44 | 494.10 | 494.37 | 109.56 | 839.44 | −3.20 | 0.32 | −5.47 | 4 |
| IIIn | 0 | 0 | 243.28 | 2.58 | 2 | 2 | 1 | 0.38 | 494.01 | 474.61 | 109.70 | 803.45 | −2.72 | 0.23 | −5.36 | 4 |
| IIIo | 1a | 0 | 304.19 | 3.03 | 2 | 2 | 1 | 0.43 | 492.35 | 497.87 | 109.72 | 845.33 | −3.29 | 0.34 | −5.52 | 3 |
| IIIp | 1a | 0 | 234.28 | 2.77 | 2 | 2 | 1 | 0.38 | 493.27 | 474.89 | 109.62 | 803.69 | −3.11 | 0.23 | −5.36 | 3 |
| IIIq | 0 | 0 | 243.28 | 2.59 | 2 | 2 | 1 | 0.37 | 495.10 | 470.47 | 104.54 | 808.04 | −2.73 | 0.24 | −5.23 | 4 |
| IIIr | 0 | 0 | 304.73 | 2.20 | 3 | 2 | 2 | −0.50 | 67.63 | 526.87 | 109.23 | 905.68 | −3.20 | 0.30 | −5.45 | 4 |
| IIIs | 0 | 0 | 334.21 | 3.10 | 3 | 2 | 2 | 0.36 | 495.03 | 535.65 | 197.04 | 926.14 | −3.58 | 0.39 | −5.46 | 5 |
| IIIt | 0 | 0 | 286.29 | 1.07 | 4 | 3 | 3 | −1.06 | 25.05 | 511.41 | 103.30 | 884.99 | −2.26 | 0.02 | −5.28 | 6 |
| IIIu | 0 | 0 | 225.29 | 2.37 | 2 | 2 | 1 | 0.27 | 496.43 | 471.63 | 108.05 | 791.71 | −2.47 | 0.34 | −5.62 | 4 |
 | ||
| Fig. 4 2D pairwise scatter plots of selected ADMET properties: (A) molecular weight against lipophilicity, (B) molecular weight against number of hydrogen bond acceptors, (C) molecular weight against dipole moments, (D) molecular weight against number of rotatable bonds, (E) molecular weight against cohesion indices, (F) molecular weight against blood–brain barrier penetration coefficient, (G) molecular weight against predicted logIC50 of HERG channel blockage, (H) molecular weight against predicted binding coefficient to human serum albumin, (I) molecular weight against skin penetration parameter, (J) molecular weight against aqueous solubility, (K) molecular weight against Caco-2 penetration, (L) molecular weight against Madin–Darby canine kidney parameter. | ||
Experimental
General experimental procedures
The synthesis of organic compounds was carried out with commercially available chemicals that were used as received (Scheme 1). All target compounds were tested in the form of the hydrochlorides which were obtained by dissolution of the corresponding free base in ethanolic HCl. Nuclear Magnetic Resonance spectroscopy data were recorded using UNITYplus – 500 spectrometer (1H NMR at 500 MHz). Chemical shifts are denoted in unit parts per million (ppm) relative to the solvent (1H NMR peak: 3.30 for CD3OD). The following abbreviations are used to indicate peak multiplicities and characteristics: s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet). Mass spectra were recorded on a BIOTO FIIEST mass spectrometer. TLC analyses were carried out on aluminum plates (Merck) precoated with silica gel 60 F254 (0.2 mm thickness). Visualization of spots was performed with UV light or by treatment with iodine.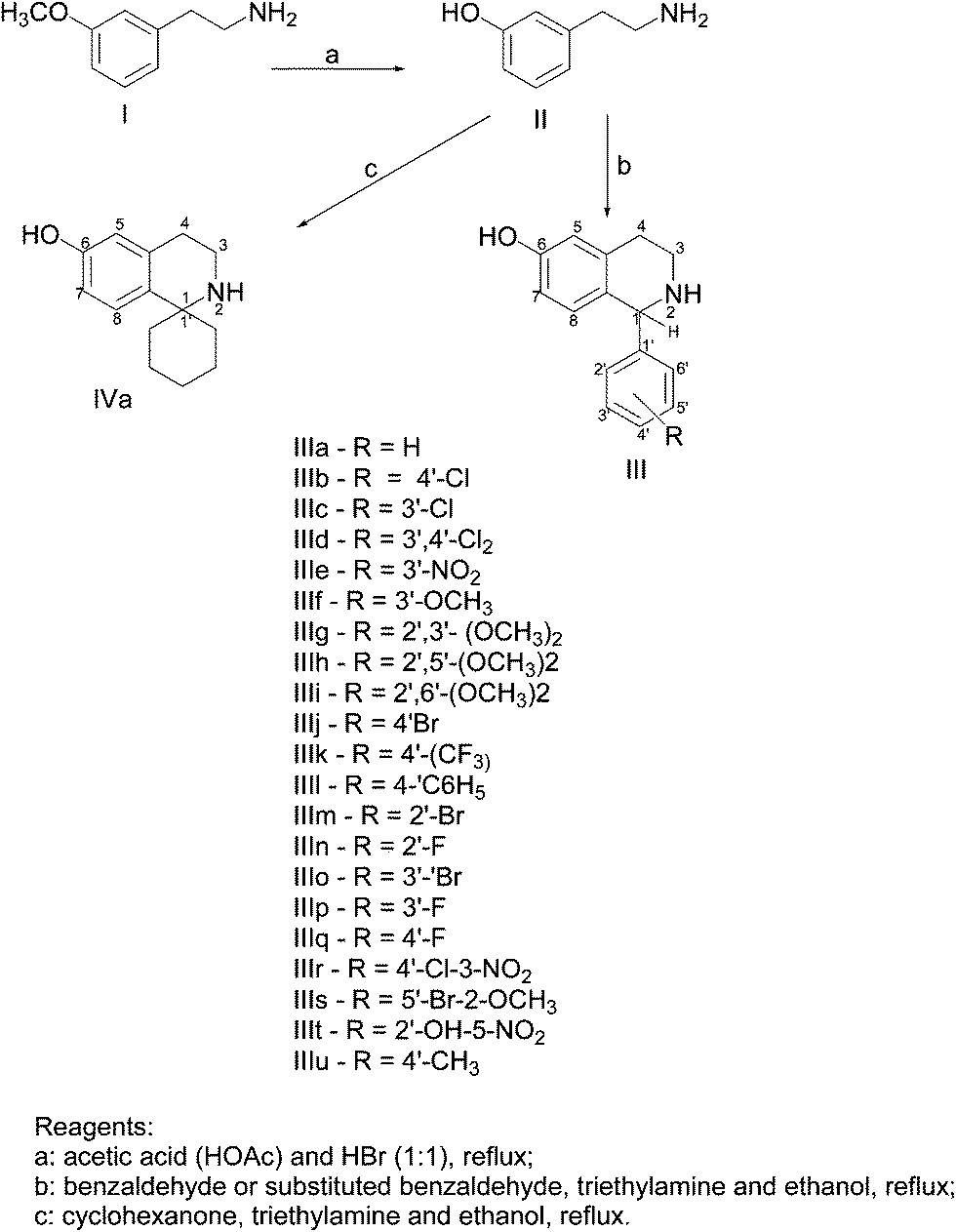 | ||
| Scheme 1 Synthesis of 6-hydroxyl-1,2,3,4-tetrahydroisoquinoline analogues. | ||
General procedure for the synthesis of 1-aryl-6-hydroxy-1,2,3,4-tetrahydroisoquinolines
In vitro sensitivity assays of P. falciparum ([3H]-hypoxanthine incorporation)
The test samples were assayed on a P. falciparum clone designated K1 which is chloroquine/pyrimethamine resistant. Test samples of 10 mg were prepared in dimethyl sulfoxide (DMSO). The testing procedure was as follows: a serial dilution factor of 1:2 was prepared with 100 μL of medium RPMI 1640 without hypoxanthine 10.44 g L−1; HEPES 5.94 g L−1; Albumax® 5 g L−1; neomycin 10 mL L−1 (100 U mL−1); NaHCO3 50 g L−1 stock 42 mL L−1 (2.1 g L−1), the final concentration was distributed into 96 wells of the Costar TM 96 – wells microtitre plates. To these 96 wells, 100 μL of medium, washed human red blood cells A+ (RBC) and P. falciparum mix, were added. The plates were incubated for 48 hours at 37 °C in a chamber and gassed with a 4% CO2, 3% O2, 93% N2. After 50 μL of medium and [3H]-hypoxanthine (0.5 μCi) were added to each well. The plates were then put back into the chamber and gassed with a 4% CO2, 3% O2, and 93% N2 mix. The chamber was then placed in the incubator for 24 hours at 37 °C. The assay was terminated by harvesting the content of each microtiter plate. Data were transferred into a graphic program (Excel) and analyzed to determine the IC50. The test scores were grouped as follows; inactive (no repeat) IC50 > 5 μg mL−1; moderate activity (repeat) 0.5 μg mL−1 < IC50 < 5 μg mL−1; high activity (repeat) IC50 < 0.5 μg mL−1.34,35
In vitro sensitivity assays (cytotoxicity)
The test samples were submitted to L-6 (rat skeletal myoblast cells). Compounds were prepared in DMSO the testing procedure was as follows. A sample of 100 μL medium (RPMI + 10% FCS + 1.7 μML – glutamine (850 μL 200 mM for 100 mL)), was added to wells of a microtiter plate. Then 100 μL of a cell suspension of 4 × 104 cells per mL was added. After a serial dilution factor of 1:3 was prepared into the wells. The plates were then incubated for 70 hours at 37 °C/5%CO2.
In silico modelling
Each compound was sketched using the GaussView software and geometry optimisation was carried out using the Gaussian 09W package36 with the density functional theory (DFT) using the B3LYP/6-31+G(d,p) approach until convergence was reached. This was necessary in order to generate good starting geometries for the 3D models. The low-energy 3D chemical structures were saved in mol2 format and initially treated with LigPrep,37 distributed by Schrodinger, Inc. This implementation was carried out with the graphical user interface (GUI) of the Maestro software package,38 using the OPLS force field.39–41 The treatment with LigPrep was necessary in order to correct protonation states and set the right parameters necessary for running QikProp.42 Protonation states at biologically relevant pH were correctly assigned (group I metals in simple salts were disconnected, strong acids were deprotonated and strong bases protonated, while explicit hydrogens were added). A set of the ADMET-related properties (a total of 46 molecular descriptors) were calculated using the QikProp program42 running in normal mode. QikProp generates physically relevant descriptors and uses them to perform ADMET predictions. An overall ADMET-compliance score, drug-likeness parameter (indicated by #stars), was used to assess the pharmacokinetic profiles of the compounds. The #stars parameter indicates the number of property descriptors computed by QikProp, which fall outside the optimum range of values for 95% of known drugs. The methods implemented were developed by Jorgensen et al.43–45 All molecular modelling was carried out on a Linux workstation with a 3.5 GHz Intel Core2 Duo processor.Conclusions
A series of 1-aryl-6-hydroxy-1,2,3,4-tetrahydroisoquinoline analogues was synthesized using simple methodology. To the best of our knowledge this is the first time the antiplasmodial of these analogues are reported. Following the criteria established by the WHO/TDR screening program,46 a compound is considered active against P. falciparum when in vitro IC50 is <1 μM (or 0.2 μg mL−1) and SI > 100. In the present study, three compounds (IIIb, IIIk and IVb) fell within this category and could be described as antimalarial hits from which more potent THIQ analogues could be obtained by further investigation. Additionally, in vivo and mechanistic studies of these compounds are underway. The study clearly demonstrates that 1-aryl-6-hydroxy-1,2,3,4-tetrahydroisoquinolines display sufficient antiplasmodial activity and favourable predicted pharmacokinetic properties to warrant further investigation of their antiplasmodial activity.Acknowledgements
The academic license is acknowledged from Schrodinger Inc., for running the QikProp software.Notes and references
- P. D. Hoeprich, in Infectious Diseases, J. B. Lippincott Company, Philadelphia, 4th edn, 1972, p. 26 Search PubMed.
- A. Fournet and V. Munoz, Curr. Top. Med. Chem., 2002, 2, 1915 CrossRef.
- J. D. Phillipson and C. W. Wright, J. Ethnopharmacol., 1991, 32, 155 CrossRef CAS.
- S. Schwikkard and F. R. van Heerden, Nat. Prod. Rep., 2002, 19, 675 RSC.
- A. Gringuaz, Antimalarial Drugs II, Chemotherapy of malaria, in Introduction to Medicinal Chemistry, Wiley-Vc Inc., New York, 1997, vol. 197, pp. 202–210 Search PubMed.
- N. J. White, J. Clin. Invest., 2004, 113, 1084 CrossRef CAS PubMed.
- S. I. Murahashi and T. Shiola, Tetrahedron Lett., 1987, 28(21), 2383 CrossRef CAS.
- N. J. O'reilly, W. S. Derwin and H. C. Lin, Synthesis, 1990, 7, 550 CrossRef.
- M. Strolin-Bendetti, P. Doistert and P. J. Cannitiati, J. Neural Transm., 1989, 78, 43 CrossRef.
- P. Yakoyama, T. Ohwada and K. J. Shudo, Org. Chem., 1999, 64, 611 CrossRef.
- G. Bringman, R. Brun, M. Kaiser and S. Neumann, Eur. J. Med. Chem., 2008, 43(1), 32 CrossRef PubMed.
- G. François, G. Timperman, W. Eling, L. A. Assi, J. Hotemz and G. Bringmann, Antimicrob. Agents Chemother., 1997, 41, 2533 Search PubMed.
- G. Bringmann, K. Messer, K. Wolf, J. Mühlbacher, M. Grüne, R. Brun and A. M. Louis, Phytochemistry, 2002, 60, 389 CrossRef CAS.
- G. Bringmann, C. Günthera, W. Saeb, J. Mies, R. Brun and L. Aké Assi, Phytochemistry, 2000, 54, 337 CrossRef CAS.
- Y. Iwasawa and A. Kiyomoto, Jpn. J. Pharmacol., 1967, 17, 143 CrossRef CAS.
- P. Cheng, N. Huang, Z.-Y. Jiang, Q. Zhang, Y.-T. Zheng, J.-J. Chen, X.-M. Zhang and Y.-B. Ma, Bioorg. Med. Chem. Lett., 2008, 18, 2475 CrossRef CAS PubMed.
- P. Manmi, M. d'Ischia and G. J. Prota, Org. Chem., 2001, 66, 5048 CrossRef PubMed.
- Z. Z. Liu and V. F. Tang, Chin. Lett., 2003, 12, 947 Search PubMed.
- J. D. Scott and R. M. Williams, Angew. Chem., Int. Ed., 2001, 40, 1463 CrossRef CAS.
- A. Kumar, B. S. Katiyar, S. Gupta and M. S. Prem, Eur. J. Med. Chem., 2002, 61(2), 195 Search PubMed.
- T. Kametani, K. Kigasawa, M. Hiiragi and H. J. Ishimaru, J. Chem. Soc. C, 1971, 2632, 10.1039/J39710002632.
- P. F. Moundipa, F. N. Njayou, S. Yantitoum, B. Sonke and F. M. Tchouanguep, Cam. J. Biol. Biochem. Sci., 2002, 12, 39 Search PubMed.
- K. Saidu, J. Onah, A. Orisadipe, A. Olusola, C. Wambebe and K. Gamaniel, J. Ethnopharmacol., 2000, 71, 275 CrossRef CAS.
- F. R. Irvine, Woody Plants of Ghana, University Press, London, Oxford, 1961, p. 358 Search PubMed.
- R. M. Soto-Hernández, R. García-Mateos, R. S. Miguel-Chávez, G. Kite, M. Martínez-Vázquez and A. C. Ramos-Valdivia, Erythrina, a Potential Source of Chemicals from the Neotropics, in Bioactive Compounds in Phytomedicine, Intech, 2012, pp. 163–184 Search PubMed.
- M. I. Aguilar, F. Giral and O. Espejo, Phytochemistry, 1981, 20, 2061 CrossRef CAS.
- H. Van de Waterbeemd and E. Gifford, Nat. Rev. Drug Discovery, 2003, 2, 192 CrossRef CAS PubMed.
- (a) C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 1997, 23, 3 CrossRef CAS; (b) C. A. Lipinski, J. Pharmacol. Toxicol. Methods, 2000, 44, 253 CrossRef.
- D. F. Veber, S. R. Johnson, H. Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, J. Med. Chem., 2002, 45, 2615 CrossRef CAS PubMed.
- P. L. Hedley, P. Jørgensen, S. Schlamowitz, R. Wangari, J. Moolman-Smook, P. A. Brink, J. K. Kanters, V. A. Corfield and M. Christiansen, Hum. Mutat., 2009, 30, 1486 CrossRef CAS PubMed.
- J. I. Vandenberg, B. D. Walker and T. J. Campbell, Trends Pharmacol. Sci., 2001, 22, 240 CrossRef CAS.
- N. Chiesa, B. Rosati, A. Arcangeli, M. Olivotto and E. Wanke, J. Physiol., 1997, 501, 31 Search PubMed.
- A. M. Aronov, Drug Discovery Today, 2005, 10, 149 CrossRef CAS.
- R. E. Desjardins, C. J. Canfield, J. D. Haynes and J. D. Chulay, Antimicrob. Agents Chemother., 1979, 16, 710 CrossRef CAS.
- J. D. Berman and J. V. Gallalee, Antimicrob. Agents Chemother., 1985, 28, 723 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria and M. A. Robb, et al., Gaussian 09, Gaussian, Inc., Pittsburgh, PA, 2009 Search PubMed.
- Schrödinger: LigPrep software, version 2.5, LLC, New York, NY, 2011 Search PubMed.
- Schrödinger: Maestro, version 9.2, LLC, New York, NY, 2011 Search PubMed.
- D. Shivakumar, J. Williams, Y. Wu, W. Damm, J. Shelley and W. Sherman, J. Chem. Theory Comput., 2010, 6, 1509 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118(45), 11225 CrossRef CAS.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110(6), 1657 CrossRef CAS.
- Schrödinger: QikProp, version 3.4, LLC, New York, NY, 2011 Search PubMed.
- W. L. Jorgensen and E. M. Duffy, Adv. Drug Delivery Rev., 2002, 54, 355 CrossRef CAS.
- E. M. Duffy and W. L. Jorgensen, J. Am. Chem. Soc., 2000, 122, 2878 CrossRef CAS.
- W. L. Jorgensen and E. M. Duffy, Bioorg. Med. Chem. Lett., 2000, 10, 1155 CrossRef CAS.
- S. Nwaka, B. Ramirez, R. Brun, R. L. Maes, F. Douglas and R. Ridley, PLoS Neglected Trop. Dis., 2009, 3, e440 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra46791k |
| ‡ Current address: Department of Chemistry, Faculty of Science, University of Douala, P. O. Box 24157, Douala, Littoral Region, Cameroon |
| This journal is © The Royal Society of Chemistry 2014 |