
Bench-scale demonstration of CO2 capture with electrochemically-mediated amine regeneration
Abstract
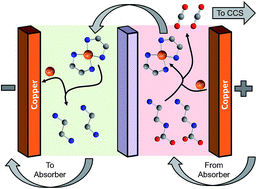
Electrochemically-Mediated Amine Regeneration (EMAR) is a new strategy for CO2 separations that uses industrially relevant amine sorbents in a nearly isothermal capture process. The electric nature of the EMAR system offers significant practical advantages in retrofit applications and for installation in non-power generating facilities. EMAR systems are based on an electrochemical copper cycle where desorption is facilitated by oxidation of a copper anode to generate cupric ions that displace CO2 from the polyamine sorbents. The CO2 capacity of the sorbent solution is regenerated through electroplating of the cupric ions onto a separate copper cathode. Results from a bench-scale system demonstrate the potential of the technology for efficient low-energy capture of CO2. The importance of current density and sorbent flow rate are measured. The work of capture increases linearly with current density. For flow rate, however, there is an optimum value of about 1 cm s−1 of superficial velocity through the device. Different electrode geometries and different electrolytes are also examined. At ambient pressure and temperature, which are far from the ideal operating conditions, CO2 can be captured for 100 kJ per mol−1 at a membrane current density of 50 A m−2. The Faradaic (current) efficiencies reach 80% under some conditions with only minimal optimization of the design and materials. This level of performance already meets or exceeds the performance of other electrochemical systems.
 Please wait while we load your content...
Please wait while we load your content...

