
Effect of polyhedral oligomeric silsesquioxane (POSS) on crystallization behaviors of POSS/polydimethylsiloxane rubber nanocomposites†
Abstract
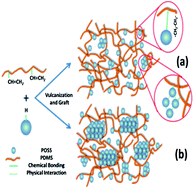
A series of heptaphenylhydrogensilsesquioxane/polydimethylsiloxane (POSS/PDMS) nanocomposites are prepared through grafting and blending. Subsequently, the melting and isothermal crystallization behaviors of the POSS/PDMS nanocomposites are investigated by differential scanning calorimetery (DSC). There is an evident fluctuation in melting temperature though the melting curves of the nanocomposites show similar crystallinities to PDMS. The results indicate that the crystallization rate of the nanocomposites increases with the addition of POSS before POSS loading reaches 3 wt%, but it decreases with further addition of POSS. The maximum crystallization rate of the nanocomposites even achieves more than 2.5 times that of neat PDMS. According to X-ray diffraction (XRD) and scanning electron microscopy (SEM) results, uniformly dispersed POSS is more efficient in behaving as a nucleating agent, while the large agglomerates of POSS tend to crystallize by themselves, whose crystal regions restrict the PDMS chain segments from forming ordered structures. When POSS crystallites are larger than 31.5 nm, the orientation of PDMS chain segments can be significantly depressed.
 Please wait while we load your content...
Please wait while we load your content...

