The metal delivery mechanism of transferrin and the role of bent metallocene metals towards anticancer activity – a theoretical exploration†
Dhurairajan Senthilnathan,
Anbarasan Kalaiselvan‡
,
Swaminathan Angeline Vedha and
Ponnambalam Venuvanalingam*
Theoretical & Computational Chemistry Laboratory, School of Chemistry, Bharathidasan University, Tiruchirappalli, 620024, Tamil Nadu, India. E-mail: venuvanalingam@yahoo.com; Fax: +91 431 2407045; Tel: +91 431 2407053
First published on 10th December 2013
Abstract
The metal delivery mechanism of the metalloenzyme transferrin and the antitumor active interaction of the metal ions, generated by the rapid hydrolysis of bent metallocene metals (Cp2MCl2; M = Ti, V, Nb, Mo), with the transferrin enzyme have been investigated. The initial and rate determining steps of the metal delivery mechanism have been identified. Comparisons are drawn between the antitumor action of various external metal ions and the role of the weak interactions that decide the site-specific decarboxylation reaction of transferrin have been investigated. Computational results reveal that the metal delivery mechanism starts by the site-specific concerted decarboxylation of transferrin leading to the thermodynamically unstable metal–oxo product and this unstable metal–oxo product delivers the metal ion to the nucleic acid via ATP. The computed reaction barriers for the decarboxylation step nicely correlate with the antitumor activity of the bent metallocenes.
Introduction
With the expanding library of anticancer drugs, many pass phase I & II clinical trials but the challenge is to find agents that can carry out site-specific drug delivery. Serum transferrins (hTf), the glycoproteins that belong to the transferrin superfamily, are chosen for drug delivery to malignant cells as they are primarily transport proteins.1–3 hTf preferentially binds to Fe3+, but is capable of binding to metals such as Al3+, Cr3+, Mn2+, Cu2+, Co2+, Ga3+, Zn2+ and Cd2+.4–8 This monomeric protein has two domains, namely the C-lobe and the N-lobe, which are similar in their mechanism of action.9,10 hTf is present on tumour cells to satisfy their high Fe demand due to the higher metabolic rate of malignant cells.11 hTf binds to Fe3+ ions using up to 30% of its available binding sites and therefore possesses ample space to bind to other metal ions or drug materials. Each transferrin carries two Fe3+ ions and this iron can be replaced by a number of other metals ions, while retaining the overall protein structure.12,13 Several experimental studies report on the binding of metal ions with transferrin; the binding strength directly correlates with the antitumor activity of the metal.14–19 Murray and Köpf-Maier have identified that the bent metallocene dihalides (Cp2MX2, M = Ti, V, Nb, Mo) have antitumor potential over a wide range of pH values.19–21 A theoretical report by Senthilnathan et al., also demonstrates the bonding and electronic structural properties of these metallocene–nucleotide complexes.22 Among them, titanocene dichloride has been recognized as the first non-platinum metal complex to enter clinical trials and is now in phase II clinical screening as an anticancer drug.23,24 Sadler and coworkers25 have demonstrated that the Ti(IV) from Cp2TiCl2 occupies the vacant Fe(III) binding site in transferrin and is subsequently released to the nucleus by ATP. They have proposed a mechanism where the hydrolysis of the bent metallocene dihalides (Cp2MX2) leads to the formation of a M4+ ion (M = Ti, V, Nb, Mo) that binds with the transferrin.26–31 The metal-bound transferrin delivers the metal ion to DNA via the ATP process.26,32,33 The ion delivery mechanism of transferrin takes place via a multi-step process involving a rate determining decarboxylation reaction, followed by a fast protonation of the active site residues, including the ligand. Therefore the rate limiting decarboxylation reaction is a key step in the metal delivery endocyclic process. A schematic representation of the cyclic metal delivery mechanism is presented in Fig. 1. The metal delivery mechanism clearly depicts that the subsequent breaking of the transferrin coordination sphere through decarboxylation forms a metal–oxo (OP) product. In order to deliver the metal ion to the nucleus, the metal–oxo product undergoes further dissociation by eliminating water. This can proceed either via a proton-coupled electron transfer (PCET) or via de-protonation. The subsequent steps of the metal delivery mechanism of transferrin still remain unknown experimentally. The completely dissociated M(IV) ion is transferred to ATP and this facilitates the transport of M(IV) to the nucleus for further interaction with DNA. After the metal delivery step, the active site residues of the transferrin enzyme are protonated with the assistance of a proton pump in the metal delivery cycle. The protonated active site residue is complexed with the M(IV) ion for further cyclic processes. The detailed schematic representation of the transferrin metal transfer pathway and the involvement of the reported antitumor active bent metallocenes is presented in Fig. 1.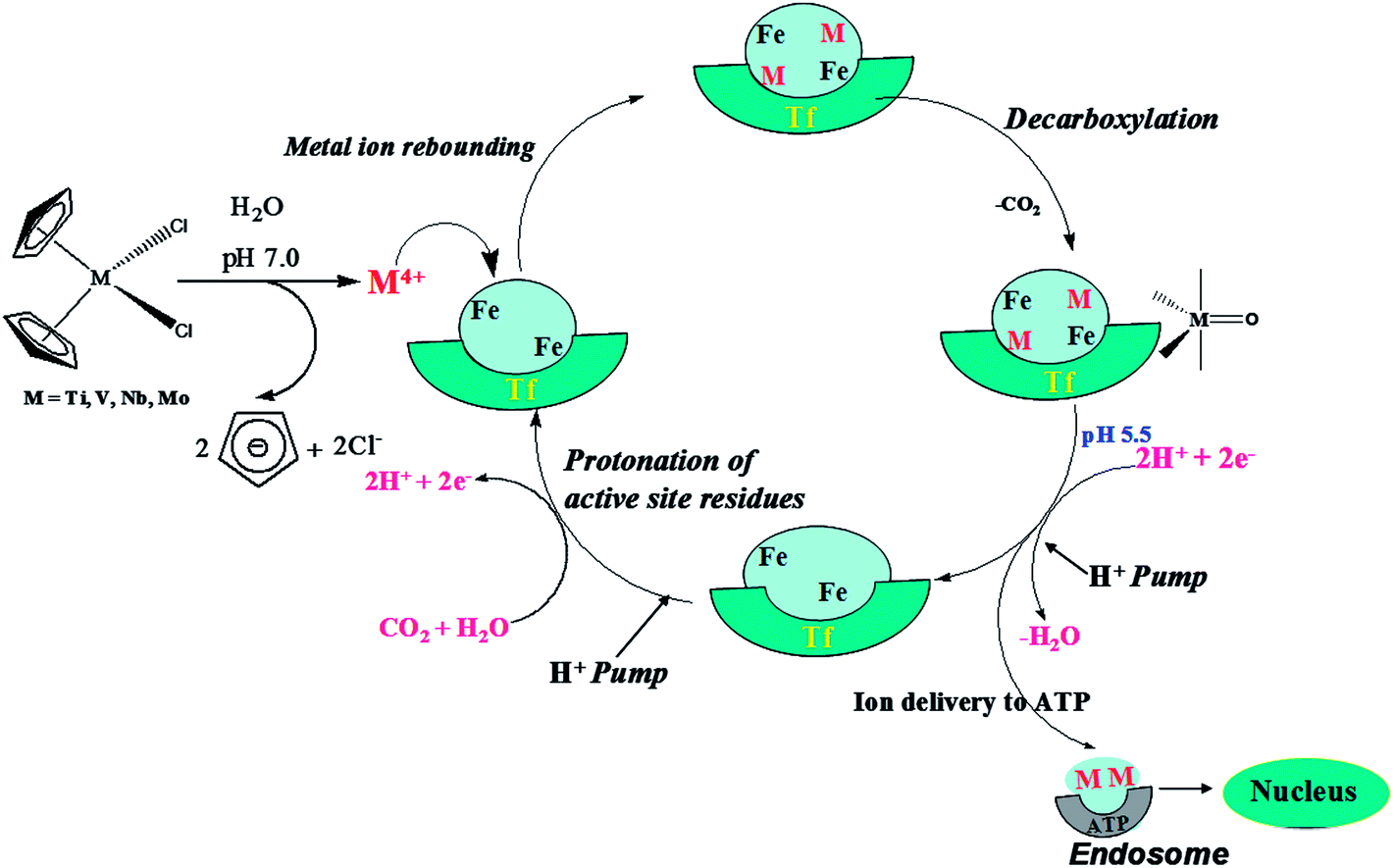 | ||
| Fig. 1 Schematic representation of the interaction of the bent metallocenes with transferrin which facilitates cell uptake and delivery of active metal ions to the cells. | ||
This work has been devoted to the identification of the mechanistic pathway for the transferrin mediated metal delivery mechanism and to investigate the role of the bent metallocene drugs in antitumor actions. The site selective decarboxylation pathway, the electronic structure analysis of the species involved and the role of the Arg124 residue on the site selective decarboxylation step have been particularly investigated.
Computational methods
Simulations have been carried out in the gas phase and solution phase to understand the nature of the mechanism, the role of the weak interactions and the enzyme action. All the geometry optimizations of the enzyme have been performed using the ONIOM34–36 method. From the ONIOM optimized geometry, the active site has been truncated and used for transition state modeling (QM) at the B3LYP/LANL2DZ level using Gaussian 03.37 The stationary points located have been characterized as either minima or a transition state (TS) by computing their harmonic vibrational frequencies. TSs have a single imaginary frequency while minima have all real frequencies. The iron(III) redox chemistry supports the octahedral complex with strong ligands showing high-spin complexes38 and the bio-coordination of Ti4+, V4+, Nb4+ and Mo4+ metal ions have a d0, d1, d1and d2 configuration, respectively. Therefore the modeling of these reactions does not include the problem of spin pairing and unpairing with metal ions. The metal delivery mechanism of transferrin has been examined by scanning the potential energy surface and monitoring the geometric changes in each reactive intermediate. All the transition states are further confirmed by intrinsic reaction coordinate analysis. Further NBO and Atoms in Molecules topological analyses have been performed and the results are discussed below.Reductionist approach
The crystal structure of human serum transferrin (hTf) with 1.6 Å resolutions has been used for this computation (ID = 1A8E).39 In this enzyme, Fe(III) and CO32− are presented as cofactors and Tyr199, Tyr95, His249 and Asp63 are presented as active site residues. The N-lobe of human serum transferrin has been considered for modeling as the metal ions in the C-lobe are rapidly replaced by Fe(III). The structure of human serum transferrin (hTf) has been taken from the Protein Data Bank. The enzyme structure has been truncated as a layer at 10 Å for the ONIOM optimization with iron as a central point35,40 and is shown in the ESI (ESI-I).† In this calculation, the important active site residues Tyr188, Tyr95, His249, Asp63 and Arg124 with iron as the cofactor have been treated quantum mechanically (QM) and the rest of the truncated section of the enzyme has been treated using a molecular mechanics (MM) method. Here the iron atom is replaced by the bent metallocene metals such as Ti, V, Nb and Mo (ESI-I†). The geometry optimizations of the transferrin enzyme have been carried out by a QM/MM approach using the ONIOM method at the B3LYP/LANL2DZ:UFF level. In other words, in the metal–transferrin (M–Tf) coordination sphere, Tyr188, Tyr95, His249, Asp63, the metal, the carbonate anion and the Arg124 residue have been treated quantum mechanically while the rest of the enzyme has been treated by a molecular mechanics method at the UFF level.41,42 Initially, OPLS and AMBER force fields were used to optimize the structure and there were some convergence difficulties in the optimization. Therefore UFF has been used as it has already been used in the optimization of metalloenzymes.43–45 The transition metal atoms have been treated using effective core potential (ECP) and the valance shells including the 3d and 4d orbitals are described by the double-ζ basis sets with diffuse 3d functions. The RMSD values in the ONIOM optimization and those of the crystal structure have been checked and are found to be almost the same (≈0.34 Å). From the QM/MM optimized transferrin, the M–Tf coordination sphere and the Arg124 residue have been used to model the cleavage. As we have employed the truncated enzyme for the QM investigation to probe the mechanism, another model has not been employed for the enzymatic site. Further analyses have been done using ADF46,47 and AIM2000.48–50Results and discussion
The electrostatic potential map of the transferrin enzyme has been calculated using Molegrow software51 and is presented in Fig. 2. The computed electrostatic potential map of the transferrin shows a positive potential around the active Fe(III) site represented by the blue color and is surrounded by a series of negative potentials (red color) corresponding to the residues Tyr188, Tyr95, His249, Asp63 and carbonate anion (Fig. 2A and B). The region of the electrostatic interaction between the cofactor and the active site residues has been presented in a crosswire circle (Fig. 2C). It is interesting to note that the negative potential of the carbonate is surrounded by the positive potential of the Arg124. This suggests a weak interaction between the Arg124 and the carbonate anion.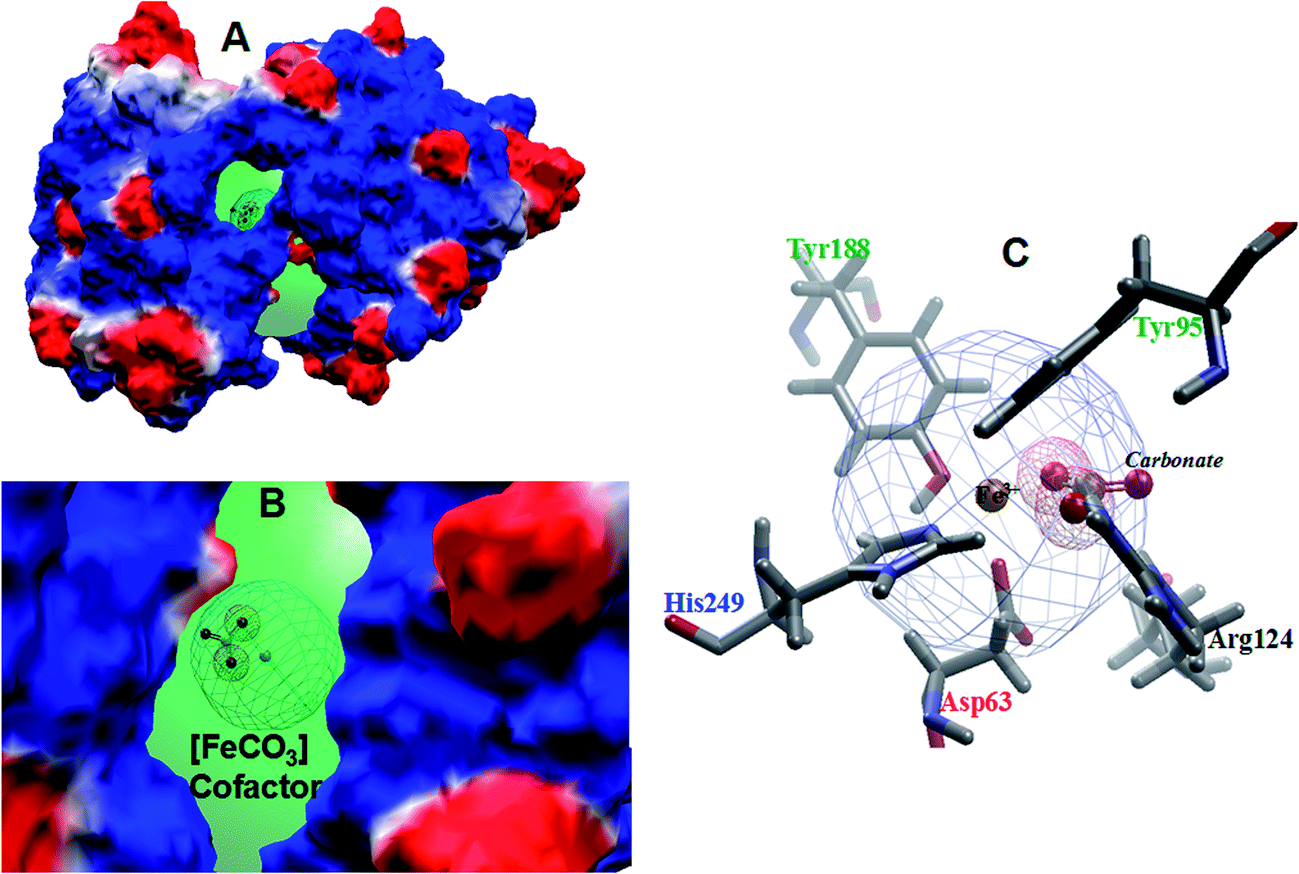 | ||
| Fig. 2 (A) The electrostatic potential surface for human serum transferrin with a cofactor, (B) the expanded view of the iron carbonate cofactor in the enzyme cavity and (C) the crystallographic view of the coordination sphere of Fe–Tf with closer active site residues. | ||
Modeling the key step of the decarboxylation reaction in the metal delivery mechanism
The key step of the metal delivery mechanism of transferrin and the possible pathways for the decarboxylation reaction followed by the metal delivery of the metal–oxo species are shown in Fig. 3.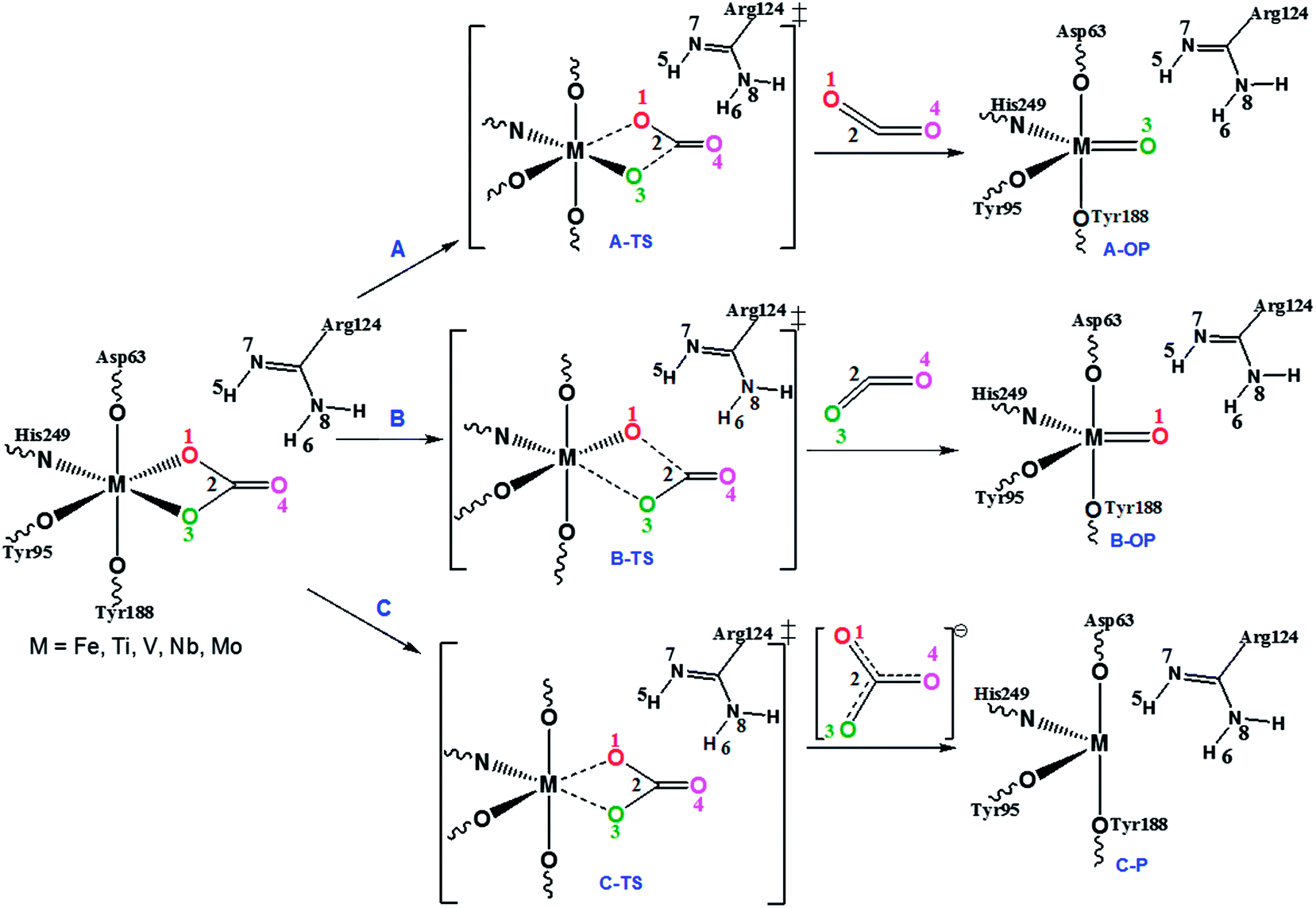 | ||
| Fig. 3 Mechanism for the transition from a closed to an open conformation of the metal–transferrin (M–Tf, M = Fe, Ti, V, Nb, Mo) by the decarboxylation reaction leading to the oxo-product. | ||
The decarboxylation reaction may take place either through (path A) breaking of the M–O1 and C2–O3 bonds (A–TS) or via (path B) breaking of the M–O3 and C2–O2 bonds (B–TS) or through (path C) the removal of CO32−. Path C is ruled out experimentally52 and therefore path C is not considered here. Path A and path B have been modeled both in the gas and solution phase.
In path A, the cleavage of the M–O1 and C2–O3 bonds takes place with the simultaneous formation of the M–O3 π-bond leading to the metal–oxo product in a concerted manner. During this reaction, the octahedral metal–transferrin coordination is converted into a distorted trigonal bipyramidal coordination. The relative free energies of the transition states and the corresponding metal–oxo products of various metal–transferrin enzymes are listed in Table 1. The activation barriers suggest that the decarboxylation reaction is faster when the metal ions of the bent metallocene bind with the transferrin enzyme and the stability of the metal–oxo product is also lower compared to the Fe(III) complex.
| M–Tf | Gas phase | Solvent phase | ||||||
|---|---|---|---|---|---|---|---|---|
| ΔG‡ | ΔGr | ΔG‡(S) | ΔGr(S) | |||||
| A–TS | B–TS | A–OP | B–OP | A–TS | B–TS | A–OP | B–OP | |
| Fe–Tf | 29.86 | 3313 | −37.87 | −37.87 | 22.96 | 24.16 | −28.18 | −28.18 |
| Ti–Tf | 11.30 | 15.38 | −7.81 | −7.81 | 7.76 | 11.35 | −13.18 | −13.18 |
| V–Tf | 15.27 | 19.27 | −16.10 | −16.10 | 12.86 | 1.98 | −18.02 | −18.02 |
| Nb–Tf | 20.82 | 24.27 | −22.43 | −22.43 | 15.50 | 19.53 | −26.63 | −26.63 |
| Mo–Tf | 23.90 | 25.89 | −23.16 | −23.16 | 16.31 | 20.18 | −27.25 | −27.25 |
The binding of Ti(IV) with the enzyme replacing Fe(III) drastically reduces the energy barrier from 29.86 kcal mol−1 to 11.30 kcal mol−1 and also decreases the stability of the metal–oxo product. The relative free energy for the reaction of Fe–oxo product is −37.87 kcal mol−1 while it is −7.81 kcal mol−1 for the Ti–oxo product. This demonstrates that the Ti–oxo product is relatively less stable and therefore it easily undergoes protonation with the help of the proton pump and delivers the Ti(IV) to the tumor cells enhances the antitumor activity.
From Table 1, it is clear that the relative order of reactivity of the M–Tf towards decarboxylation with various metals is as follows:
| Fe–Tf < Mo–Tf < Nb–Tf < V–Tf < Ti–Tf. |
Metal–oxo species dissociation pathways
The metal delivery mechanism of transferrin consists of several individual biological steps as reported so far. According to earlier reports, the key step is the decarboxylation reaction (path B) which is highly favorable, and the final product is the thermodynamically less stable metal–oxo product (B–OP). Several earlier reports suggest that the complexed metal has to transfer from the endocytosis region to the exocytosis region in the nucleus using the transferrin enzyme.53,54 The general schematic representation of the metal delivery to ATP from the thermodynamically less stable metal–oxo species is presented in Fig. 4. Biologically, there are two different metal delivery pathways that are well known; one is the proton-coupled electron transfer (PCET) metal delivery step and the other is protonation followed by the metal delivery step.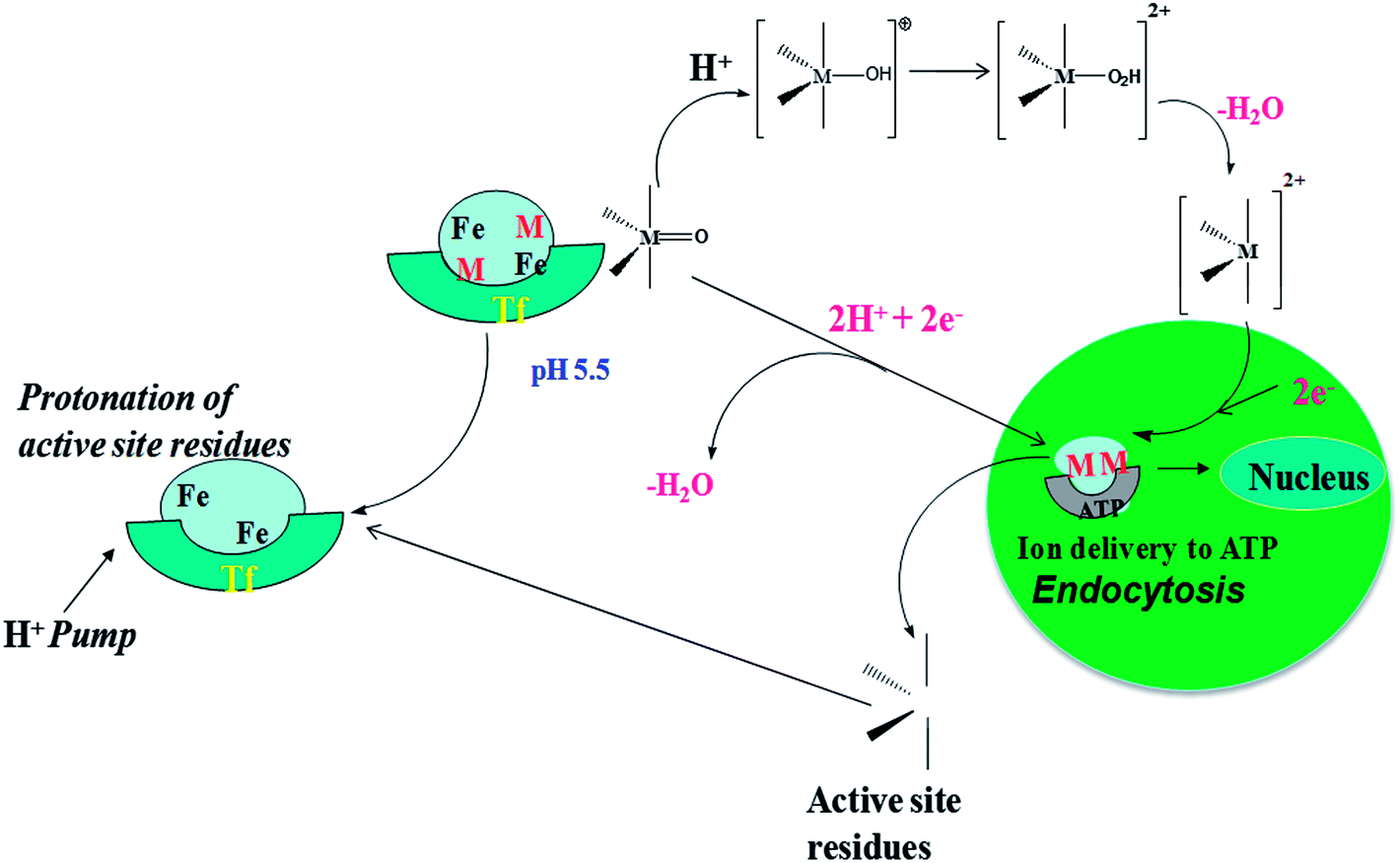 | ||
| Fig. 4 Schematic representation of the metal delivery step for the metal–oxo product in the endocytosis region. | ||
The schematic representation of the detailed metal delivery step is presented in Fig. 5. The PCET followed by the elimination of water is represented as path D. Path D consists of a mono-PCET product (1), bi-proton-coupled electron transfer product (2) and water eliminated product (3).
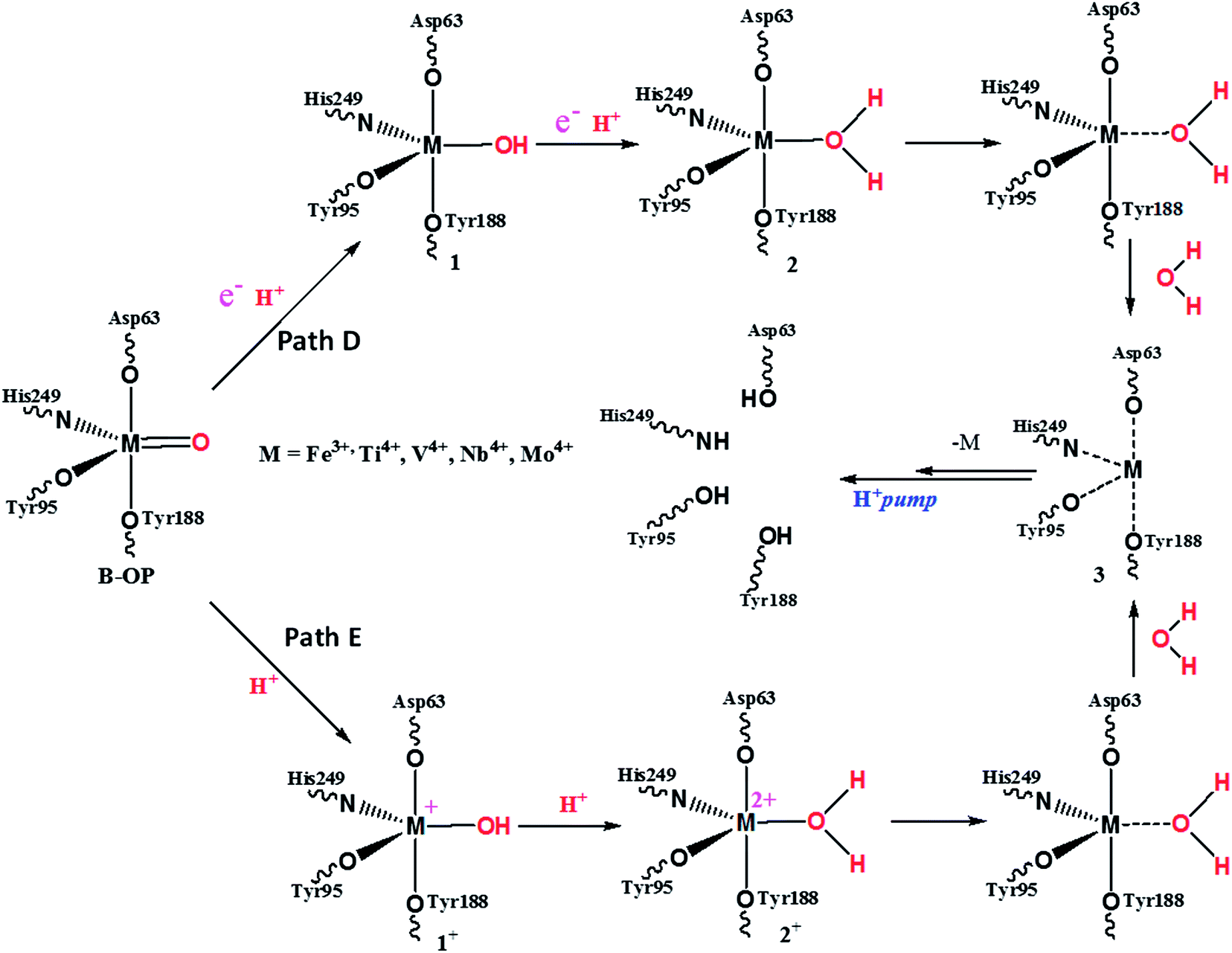 | ||
| Fig. 5 Schematic representation of the metal delivery step of the metal (M)–oxo product (B–OP) (M–Tf, M = Fe, Ti, V, Nb, Mo). | ||
The acidic protonated metal delivery pathway has been represented as path E and this pathway consists of a mono-protonation product (1+), bi-protonated product (2+) and water eliminated product (3). Product 3 is the same for both pathways. Product 3 is rapidly protonated and delivers the metal ion in the exocytosis region immediately. The relative energy of each species is presented in Table 2, the non-protonated metal–oxo species (B–OP) has been considered as zero and the energy of other species are presented relative to it.
| B–OP | B–OP | 1 | 1+ | 2 | 2+ | 3 |
|---|---|---|---|---|---|---|
| Fe3+ | 0.00 | 0.98 | −1.01 | 2.01 | −1.35 | −1.50 |
| Ti4+ | 0.00 | 1.02 | −1.34 | 2.35 | −1.56 | −1.22 |
| V4+ | 0.00 | 0.96 | −1.03 | 1.99 | −1.29 | −1.34 |
| Nb4+ | 0.00 | 0.99 | −1.12 | 2.14 | −1.43 | −1.41 |
| Mo4+ | 0.00 | 0.97 | −1.02 | 2.03 | −1.39 | −1.47 |
The tabulated relative energy values and small energy differences between pathway E and pathway D indicate that there is a slight preference for the reaction mechanism to follow pathway E instead of pathway D for the metal delivery step.
Effect of the metalloenzyme geometries on the metal delivery mechanism
The thermodynamic instability of the metal–oxo product is further explained by crystal field splitting analysis. The computed crystal field splitting of the d orbitals of the V(IV) ion with the active site residues forms an octahedral geometry, however the geometry changes to a distorted trigonal bipyramidal geometry in the metal–oxo product, as shown in Fig. 6. Here the symmetry of the coordination sphere is lowered due to the removal of one of the coordination sites from the coordination sphere by the decarboxylation reaction.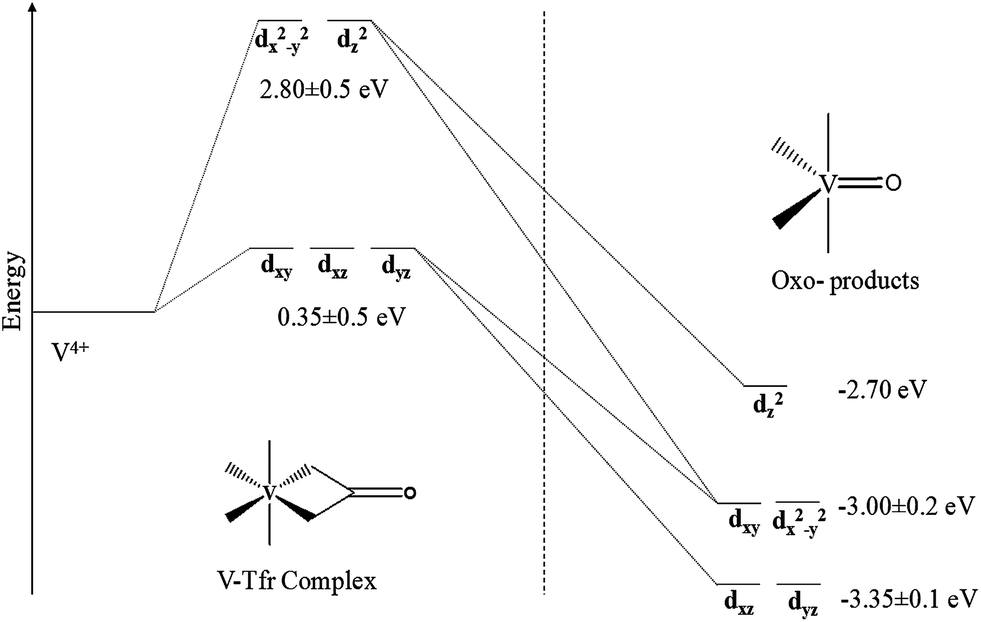 | ||
| Fig. 6 Crystal field splitting of the d orbitals of the V4+ ion in the pseudo-octahedral environment changing to a distorted trigonal bipyramidal environment computed at BP86/TZP level. | ||
The relative reaction free energies of the metal–oxo products listed in Table 1 show that the stabilities are in the following order,
| Fe–OP > Mo–OP > Nb–OP > V–OP > Ti–OP |
The stability of the Fe– and Mo–oxo products are relatively higher than the other metal–oxo products (Nb–OP, V–OP and Ti–OP). This is due to the fact that, the Fe and Mo coordination spheres are highly saturated and trigonal bipyramidal geometries of these oxo species have been observed experimentally,52 while trigonal bipyramidal geometries of the others are not known. In the case of the Ti–OP coordination sphere, it is highly unsaturated and is deformed easily, therefore the stability of the Ti–OP is very low. The effect of water as a solvent on the decarboxylation reaction has been examined through PCM calculations. The solvent effects lower the activation barriers uniformly and slightly stabilize the oxo-products but the same trend in the reactivity is predicted.
Importance of the Arg124 hydrogen bonds with the cofactor
According to the electrostatic potential map, the Arg124 amino acid residue is located close to the carbonate cofactor. In order to explain the existing hydrogen bonding interactions between Arg124 and metal ion carbonate cofactor, the NBO55 analysis and AIM topological analysis have been carried out for the transferrin complexes (M–Tf; M = Fe, Ti, V, Nb, Mo) and the NBO results are presented in Table 3.| M–Tf | LP(O1) → σ* (H5–N7) | LP(O4) → σ* (H6–N8) |
|---|---|---|
| Fe–Tf | 10.16 | 12.47 |
| Ti–Tf | 14.67 | 21.82 |
| V–Tf | 11.83 | 21.76 |
| Nb–Tf | 10.89 | 18.29 |
| Mo–Tf | 10.71 | 17.63 |
The second order perturbation energies nicely explain the interaction between the lone pair of the carbonate cofactor oxygen and the σ* of the N–H bond on the Arg124 residues. The lone pair electron density of the O1 and O4 is transferred to the σ* of the H5–N7 and H6–N8 bond respectively. According to the calculated interaction energy values the latter interaction is stronger than the former. These hydrogen bonding interactions create an eight atom cyclic hydrogen bonding network.
Topological analysis
The critical points and molecular graph of the low lying transition states of the decarboxylation and the trajectory of the gradient vector for the electron density have been calculated and presented in the ESI (Fig. ESI-S3†). The ring critical point (RCP) has been observed in all the transition state planes of bent metallocene complexes and this confirms the concerted mechanism of the decarboxylation reaction. In the transition state, the R22(8) motif hydrogen bonding network is broken and the Arg124 residues slightly move away from the original place. This dynamical movement of the Arg124 and concerted breaking of M–O1 and O3–C2 offer the decarboxylated metal–oxo product with the necessary change in coordination. In the transition state, the weak interaction is also observed with a very low negative Laplacian (−0.0004) between the Arg124 residue and the O1 atom, this value explains that the released CO2 is not inhibited by the Arg124 residue. The gradient plot of the electron density of the transition state of all the bent metallocene–transferrin complexes is calculated and presented in the ESI (Fig. ESI-S3†). The electron density distribution in the transition state of the decarboxylation reaction and observation of the ring critical point in the transition state plane evidently explain the concerted decarboxylation and the site specificity of the decarboxylation reaction in the transferrin metal delivery mechanism.The computed molecular graph shows the critical points of the transferrin coordinated with the active site residues and is presented in the ESI (figure ESI-S2).† The trajectory of the gradient vector field of the electron density at the hydrogen bonding network plane and the expanded view of gradient map are presented in Fig. 7.
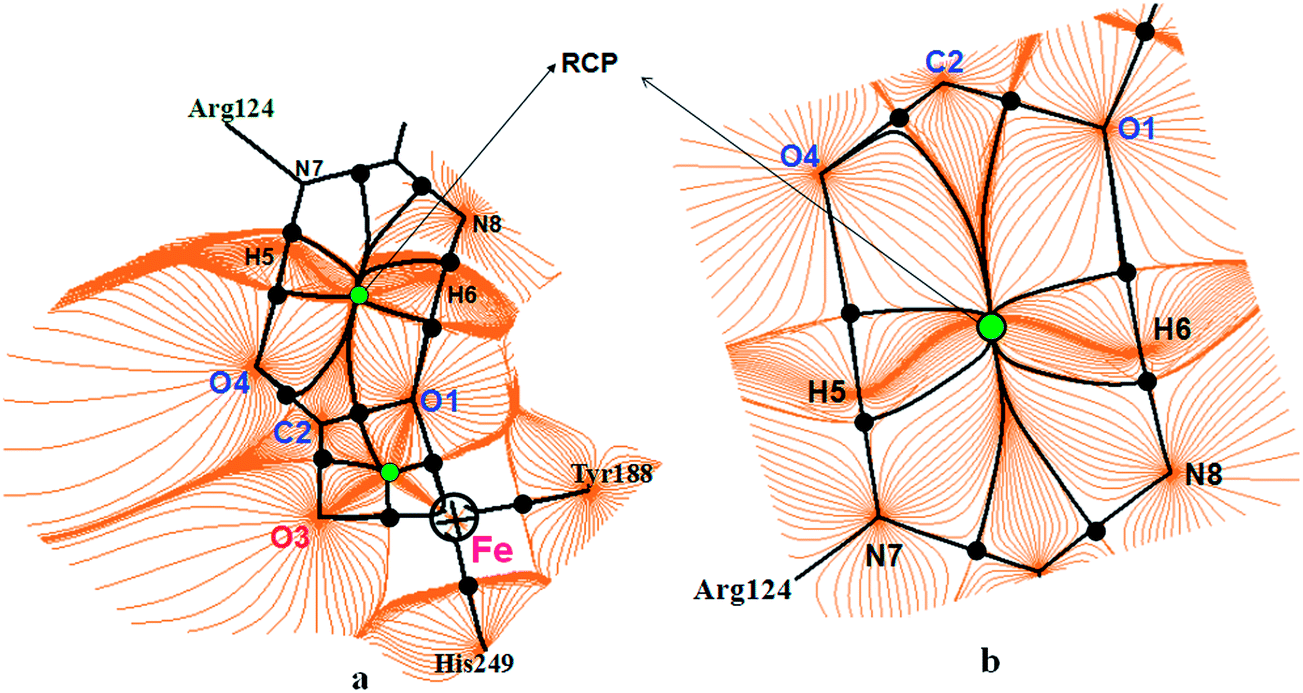 | ||
| Fig. 7 Gradient trajectories mapped on a total electron density plot in the hydrogen bonding network of the Arg124 residue with the bio-coordinated transferrin enzyme cofactor [FeCO3], showing the atom basins, bond paths (black lines), bond critical points (black solid circles), and a ring critical point (green solid circle). | ||
The carbonate cofactor of the transferrin and Arg124 hydrogens are located nearly in the same plane and they are also visible in the trajectory plot, including their associated atomic basins, BCPs, and bond paths. The Arg124 hydrogens make two hydrogen bonds with the O1 and O4 cofactor oxygens through their H6–N8 and H5–N7 bonds, respectively (Fig. 7). The Laplacian values at the bond critical point of the O1⋯H5 hydrogen bond (−0.0331 au) and the bond critical point of the O4⋯H6 bond (−0.0330 au) are observed and these negative Laplacian values (−∇2ρ) confirm the hydrogen bonding interaction. The gradient vector field of the electron density of the observed hydrogen bonding network of the transferrin bio-coordination explained by the zero flux of the density gradient along the bond critical points further supports this hydrogen bonding interaction.
The obtained results reveal that the low lying pathway for the decarboxylation step leads to the thermodynamically less stable distorted trigonal bipyramidal metal–oxo product.
Conclusions
In conclusion, the biologically complex and mechanistically important iron transport mechanism and decarboxylation reaction of several metal–transferrin (M–Tf, where M = Ti, V, Nb, Mo and Fe) complexes have been modeled at the B3LYP/LANL2DZ level. The topological analysis and electronic structure investigations have been carried out with a view to explore the relative antitumor activity of bent metallocene metals and to gain insight into the metal delivery mechanism involved in antitumor action. Computations reveal that (i) the binding strengths of the metals considered for the study with the transferrin enzyme are comparatively higher than that of Fe(III) metal and the activity of the transferrin is increased when the bent metallocene metals bind on the vacant sites; the increase in activity is in the following order: Ti > V > Nb > Mo. (ii) The decarboxylation reaction which is responsible for the metal ion delivery to the nucleus is kinetically controlled and the order of reactivity is Ti–Tf > V–Tf > Nb–Tf > Mo–Tf > Fe–Tf. (iii) The lower the stability of the oxo product, the easier the delivery of the metal ions will be to the ATP receptor. (iv) The decarboxylation is assisted by the weak hydrogen bond between the Arg124 residue and the oxygens on the carbonate anion. The computed second order perturbation energy and the electrostatic potential map clearly explain the presence of the weak hydrogen bond interaction between the carbonate cofactor and the Arg124. (v) In this reaction, the pseudo-octahedral metal–transferrin coordination sphere is converted into a trigonal bipyramidal metal–oxo product by symmetry lowering. (vi) The solvent effect predicts the same trend in the antitumor activity. Computed results are in excellent agreement with experimental reports.Acknowledgements
We thank the Council of Scientific and Industrial Research, India, for their financial support to PV in the form of a research grant (ref. no. 02(2158)/07/EMR-II) and for the Senior Research Fellowship to DS. PV thanks the Department of Science & Technology (DST), India for the Major Research Project (ref. no: SB/S1/PC-52/2012).References
- K. Cho, X. Wang, S. Nie and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- T. R. Daniels, T. Delgado, G. Helguera and M. L. Penichet, Clin. Immunol., 2006, 121, 159–176 CrossRef CAS PubMed.
- H. Li, H. Sun and Z. M. Qian, Trends Pharmacol. Sci., 2002, 23, 206–209 CrossRef CAS.
- W. R. Harris, J. Inorg. Biochem., 1986, 27, 41–52 CrossRef CAS.
- C. K. Luk, Biochemistry, 1971, 10, 2838–2843 CrossRef CAS.
- H. Sun, M. Cox, H. Li and P. Sadler, Struct. Bonding, 1997, 7, 71–102 CrossRef.
- G. A. Trapp, Life Sci., 1983, 33, 311–316 CrossRef CAS.
- I. S. Trowbridge and F. Lopez, Proc. Natl. Acad. Sci. U. S. A., 1982, 79, 1175 CrossRef CAS.
- T. R. Daniels, T. Delgado, J. A. Rodriguez, G. Helguera and M. L. Penichet, Proc. Natl. Acad. Sci. U. S. A., 2006, 121, 144–158 CAS.
- A. Widera, F. Norouziyan and W. C. Shen, Adv. Drug Delivery Rev., 2003, 55, 1439–1466 CrossRef CAS PubMed.
- W. H. Tong and T. A. Rouault, BioMetals, 2007, 20, 549–564 CrossRef CAS PubMed.
- J. C. Dewan, B. Mikami, M. Hirose and J. C. Sacchettini, Biochemistry, 1993, 32, 11963–11968 CrossRef CAS.
- J. A. Lebrón, M. J. Bennett, D. E. Vaughn, A. J. Chirino, P. M. Snow, G. A. Mintier, J. N. Feder and P. J. Bjorkman, Cell, 1998, 93, 111–123 CrossRef.
- A. D. Tinoco and A. M. Valentine, J. Am. Chem. Soc., 2005, 127, 11218–11219 CrossRef CAS PubMed.
- P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 3474–3482 RSC.
- L. Y. Kuo, M. G. Kanatzidis, M. Sabat, A. L. Tipton and T. J. Marks, J. Am. Chem. Soc., 1991, 113, 9027–9045 CrossRef CAS.
- H. Sun, H. Li, R. A. Weir and P. J. Sadler, Angew. Chem., Int. Ed., 1998, 37, 1577–1579 CrossRef CAS.
- M. M. Harding and G. Mokdsi, Curr. Med. Chem., 2000, 7, 1289–1303 CrossRef CAS.
- J. H. Murray and M. M. Harding, J. Med. Chem., 1994, 37, 1936–1941 CrossRef CAS.
- P. Köpf-Maier, C. Janiak and H. Schumann, J. Cancer Res. Clin. Oncol., 1988, 114, 502–506 CrossRef.
- P. Köpf-Maier, H. Köpf and E. Neuse, J. Cancer Res. Clin. Oncol., 1984, 108, 336–340 CrossRef.
- D. Senthilnathan, S. Vaideeswaran, P. Venuvanalingam and G. Frenking, J. Mol. Model., 2011, 17, 465–475 CrossRef CAS PubMed.
- G. Lümmen, H. Sperling, H. Luboldt, T. Otto and H. Rübben, Cancer Chemother. Pharmacol., 1998, 42, 415–417 CrossRef.
- N. Kröger, U. Kleeberg, K. Mross, L. Edler and D. Hossfeld, Onkologie, 2000, 23, 60–62 CrossRef PubMed.
- H. Li, P. J. Sadler and H. Sun, Eur. J. Biochem., 1996, 242, 387–393 CAS.
- M. Guo, H. Sun, H. J. McArdle, L. Gambling and P. J. Sadler, Biochemistry, 2000, 39, 10023–10033 CrossRef CAS PubMed.
- E. Garribba, G. Micera, A. Panzanelli and D. Sanna, Inorg. Chem., 2003, 42, 3981–3987 CrossRef CAS PubMed.
- J. B. Waern, C. T. Dillon and M. M. Harding, J. Med. Chem., 2005, 48, 2093–2099 CrossRef CAS PubMed.
- M. Hémadi, N. T. Ha-Duong and J. M. El Hage Chahine, J. Mol. Biol., 2006, 358, 1125–1136 CrossRef PubMed.
- F. B. Abdallah and J. M. El Hage Chahine, J. Mol. Biol., 2000, 303, 255–266 CrossRef CAS PubMed.
- A. D. Tinoco, E. V. Eames and A. M. Valentine, J. Am. Chem. Soc., 2008, 130, 2262–2270 CrossRef CAS PubMed.
- H. Kozlowski, A. Janicka-Klos, J. Brasun, E. Gaggelli, D. Valensin and G. Valensin, Coord. Chem. Rev., 2009, 253, 2665–2685 CrossRef CAS PubMed.
- M. J. Petris, J. Mercer, J. G. Culvenor, P. Lockhart, P. Gleeson and J. Camakaris, EMBO J., 1996, 15, 6084 CAS.
- S. Humbel, S. Sieber and K. Morokuma, J. Chem. Phys., 1996, 105, 1959 CrossRef CAS PubMed.
- M. Svensson, S. Humbel, R. D. J. Froese, T. Matsubara, S. Sieber and K. Morokuma, J. Phys. Chem., 1996, 100, 19357–19363 CrossRef CAS.
- T. Vreven and K. Morokuma, J. Comput. Chem., 2000, 21, 1419–1432 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 03, revision E.01, Gaussian, Inc., Wallingford, CT, 2004 Search PubMed.
- J. Pierre, M. Fontecave and R. Crichton, BioMetals, 2002, 15, 341–346 CrossRef CAS.
- P. D. Jeffrey, M. C. Bewley, R. T. A. MacGillivray, A. B. Mason, R. C. Woodworth and E. N. Baker, Biochemistry, 1998, 37, 13978–13986 CrossRef CAS PubMed.
- T. Vreven, C. Morokuma, Ö. Farkas, H. B. Schlegel and M. J. Frisch, J. Comput. Chem., 2003, 24, 760–769 CrossRef CAS PubMed.
- S. Kasuriya, S. Namuangruk, P. Treesukol, M. Tirtowidjojo and J. Limtrakul, J. Catal., 2003, 219, 320–328 CrossRef CAS.
- G. Ujaque and F. Maseras, Principles and Applications of Density Functional Theory in Inorganic Chemistry I, 2004, pp. 51–79 Search PubMed.
- L. Xin, T. Feng, F. Yabing, X. Xin, W. Nanqin and Z. Qianer, Nano Lett., 2002, 2, 1325–1327 CrossRef.
- Z. Fabio, Q. Luca, S. Giuseppe, Z. Theodora, B. Olivier, S. Blagoj, C. S. Marcus and Y. B. Anna, J. Med. Chem., 2013, 56, 6719–6731 CrossRef PubMed.
- S. Eduardo, G. Alba, L. José, M. Marta and P. Pablo, J. Am. Chem. Soc., 2010, 132, 9111–9121 CrossRef PubMed.
- G. Te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- C. Fonseca Guerra, J. Snijders, G. Te Velde and E. Baerends, Theor. Chem. Acc., 1998, 99, 391–403 Search PubMed.
- R. G. Parr and W. Yang, Density-functional theory of atoms and molecules, Oxford University Press, USA, 1994 Search PubMed.
- F. Biegler-König and J. Schönbohm, J. Comput. Chem., 2002, 23, 1489–1494 CrossRef PubMed.
- J. Henn, D. Ilge, D. Leusser, D. Stalke and B. Engels, J. Phys. Chem. A, 2004, 108, 9442–9452 CrossRef CAS.
- N. Singh and K. Misra, Bioinformation, 2009, 3, 255 CrossRef.
- A. Ghosh, JBIC, J. Biol. Inorg. Chem., 2006, 11, 712–724 CrossRef CAS PubMed.
- M. Hémadi, P. H. Kahn, G. Miquel and J. M. E. H. Chahine, Biochemistry, 2004, 43, 1736–1745 CrossRef PubMed.
- A. N. Steere, B. F. Miller, S. E. Roberts, S. L. Byrne, N. D. Chasteen, V. C. Smith, R. T. A. MacGillivary and A. B. Mason, Biochemistry, 2012, 51, 686–694 CrossRef CAS PubMed.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: The detailed computational approach, essential bond lengths of the reactants, transitions states & products, molecular graphs of the enzyme complexes and the total electron density plots of the transition states are given. See DOI: 10.1039/c3ra46422a |
| ‡ Present address: Department of Chemistry, Anna University of Technology, Tiruchirappalli-610204, India |
| This journal is © The Royal Society of Chemistry 2014 |