Dispersion of rGO in polymeric matrices by thermodynamically favorable self-assembly of GO at oil–water interfaces†
Saeed Zajforoushan Moghaddam,
Sina Sabury and
Farhad Sharif*
Department of Polymer Engineering and Color Technology, Amirkabir University of Technology, 424 Hafez Ave, Tehran, Iran. E-mail: sharif@aut.ac.ir; Fax: +98-21-66469162; Tel: +98-21-64542409
First published on 29th November 2013
Abstract
Various approaches have been employed to disperse graphene in polymers. We introduce a new method which takes advantage of the amphiphilic nature of graphene oxide (GO). This feature results in the spontaneous self-assembly of monolayers at the oil–water interface. Thermodynamically favorable adsorption of GO sheets on the liquid droplets serves as the first step in the fabrication of well-dispersed composites. It eliminates the need for sonication and further compounding, which can destroy the structure of the graphene layers. Another equally important consideration is how to vaporize the liquid medium to preserve appropriate dispersion in the dried masterbatch. Microwave radiation is employed to vaporize the liquids and also to partially reduce GO while keeping it well-dispersed in the polymer. As a challenging matrix, the dispersion of GO in natural rubber is studied here. XRD and SEM results confirm appropriate dispersion of the particles. Improvements in the modulus and ultimate strength were observed without any significant reduction in elongation at break.
Introduction
It is common to improve the physical and mechanical properties of polymers by the incorporation of different fillers.1–4 Particulate fillers such as carbon black (CB) have been used in the reinforcement of rubbers for years. However, the need for including considerable amounts of filler to observe a significant enhancement in the mechanical properties has been a serious drawback. Recently, nanoparticles such as nanoclays and carbon nanotubes (CNTs) have been introduced as alternatives with superior specific surface areas compared with particulate fillers.1,3 Nevertheless, obstacles such as inadequate mechanical reinforcement by nanoclays and the high price of CNTs have hindered their widespread use.1The introduction of graphene in 2004 provided a new opportunity to fabricate reinforced composites.5 Graphene consists of atomically thin nanoplatelets, which are composed of carbon atoms in a honeycomb structure. It has been reported6 to theoretically have a Young's modulus of 1 TPa and an ultimate strength of about 130 GPa, making it the stiffest monolayer found to date. This extraordinary mechanical performance is due to the perfect in-plane structure of carbon atoms, held together by strong covalent bonds. Moreover, because of the remarkable specific surface area (theoretically about 2300 m2 g−1), it is conceivable that strong interfacial bonding with polymeric matrices can be established.1,3 Therefore, higher improvement in properties with lower content of filler is expected.
The primary prerequisite in the fabrication of graphene composites is to obtain a uniform and homogenous dispersion of monolayers in the polymeric matrix.1–4,7,8 Mainly, graphene composites can be fabricated by four methods: (i) direct mixing, (ii) solution mixing, (iii) in situ polymerization and (iv) latex mixing.1,3 The dispersion of graphene in elastomeric matrices is more challenging due to the intrinsic difficulties of elastomer compounding.9,10 To date, the best known method to fabricate elastomer–graphene composites is the so called “latex method”.11,12 It is based on mixing an aqueous dispersion of graphene oxide and an elastomeric latex by sonication and separating the masterbatch through evaporation or coagulation by the addition of an appropriate non-solvent. This method has been employed for the production of various elastomeric composites such as NR, NBR and SBR.13–17 Nevertheless, it requires sonication for a long time, while partial flocculation is expected to occur during the segregation step.17
Recently, Pickering emulsion polymerization has been utilized to fabricate composites of polymers such as PMMA, and PS.18–21 In this case, besides the reinforcing role, the nanofiller acts as an emulsifier to stabilize the monomeric phase in the aqueous medium. For this purpose, partial wettability of the particles by both hydrophilic and hydrophobic phases is required, which is known as amphiphilic behavior.13,19 There are two main steps for the in situ polymerization. First, sonication in the presence of amphiphilic particles, which results in emulsified oily monomer droplets. Second, promoting polymerization in each emulsion droplet forms polymer particles covered by self-assembled nanofillers. Applications of the self-assembly of graphene and graphene oxide layers, including fabricating composites, are reported by several authors.22–28
Graphene oxide (GO) is a widely used precursor of graphene, which exhibits amphiphilic behavior because of its chemical and structural characteristics.29–31 The amphiphilicity of GO is due to the presence of hydrophilic groups, specifically carboxyls located at the edges, and hydrophobic aromatic regions in the center of the nanosheets.32 Based on this chemically heterogeneous structure, GO is capable of stabilizing immiscible water–oil mixtures, i.e. water–toluene, and forming stable Pickering emulsions.18,32,33
In this study, a novel method to fabricate well-dispersed reduced-graphene oxide (rGO) composites using the Pickering emulsion approach is presented. Considering the difficulties of dispersing rGO in elastomers, NR has been selected as a typical polymeric matrix. A detailed study of GO as an amphiphilic Janus nanoparticle and the corresponding Pickering emulsion is illustrated. It is shown that the amphiphilic nature of GO promptly results in the self-assembly of layers at the oil–water interface. Therefore, adsorption of GO to the polymeric phase occurs almost spontaneously, eliminating the need for long sonication and compounding times.
Experimental section
Materials
For the synthesis of GO, natural graphite flakes were supplied by Asbury Graphite Mills, USA. Concentrated sulfuric acid (H2SO4 98%), sodium nitrate (NaNO3), potassium permanganate (KMnO4 99.9%), hydrogen peroxide (H2O2 30%) and hydrochloric acid (HCl 37%) were all purchased from Merck (Darmstadt, Germany). For the compounding and vulcanization processes, toluene (chromatography grade) was purchased from Merck (Darmstadt, Germany), natural rubber (SMR 20), zinc oxide (ZnO), stearic acid (SA), sulfur (S), CZ and MBT were used as-received.Synthesis of graphene oxide (GO)
The oxidation of pristine graphite was carried out by a modified Hummers method.34 Natural graphite flakes (10 g) were added to a mixture of concentrated H2SO4 (250 cm3) and NaNO3 (5 g) and stirred by a magnetic stirrer. After a while, KMnO4 (25 g) was added to the mixture gradually over one hour. The mixture was then stirred for 24 hours at room temperature to complete the oxidation step. During this step, the mixture's viscosity increased considerably, indicating a high level of intercalation. To terminate the oxidation process, H2O2 aqueous solution was added to the mixture, until the colour changed to bright yellow. The prepared graphite oxide was then washed and filtered several times with 1 M HCl aqueous solution and deionized water through a funnel, to remove impurities and ions. The washed graphite oxide was sonicated by an ultrasonic bath sonicator (CD-4820 40W) to exfoliate the layers. Subsequently, the mixture was repeatedly centrifuged at 4500 rpm for 10 minutes and the stable dispersion was separated from the precipitates. This procedure was repeated several times until no noticeable amount of precipitated material was observed. The prepared dispersion was stable for more than 3 months.Preparation of the masterbatch
Firstly, natural rubber was dissolved in toluene by stirring for 2 days at 70 °C. Later, the bright solution was filtered and centrifuged at 4500 rpm for 5 minutes to separate precipitates and undissolved rubber. The prepared homogeneous solution was stable over 3 months without any precipitation or change in concentration. The solution was then added to deionized water in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 volumetric ratio (the ratio was selected to ensure formation of an o/w emulsion) resulting in a biphasic oil-in-water system. Later, the aqueous dispersion of GO was added to the biphasic system gradually, and the system was churned to form oil droplets in the aqueous phase. With increasing GO concentration, the viscosity of the Pickering emulsion rose sharply, finally turning into a paste. The paste-like mixture was then heated in a microwave apparatus to evaporate the water and toluene, resulting in a highly concentrated masterbatch. The prepared compound was then heated in a vacuum oven at 60 °C and 50 mbar for 48 hours to vaporize residual solvents and volatiles.
:6 volumetric ratio (the ratio was selected to ensure formation of an o/w emulsion) resulting in a biphasic oil-in-water system. Later, the aqueous dispersion of GO was added to the biphasic system gradually, and the system was churned to form oil droplets in the aqueous phase. With increasing GO concentration, the viscosity of the Pickering emulsion rose sharply, finally turning into a paste. The paste-like mixture was then heated in a microwave apparatus to evaporate the water and toluene, resulting in a highly concentrated masterbatch. The prepared compound was then heated in a vacuum oven at 60 °C and 50 mbar for 48 hours to vaporize residual solvents and volatiles.
Fabrication of NR–rGO composite
The masterbatch was compounded with raw natural rubber and other additives including curing agents using an open two-roll mill at room temperature for 15 minutes. Details of the curing formulation are given in Table 1. The vulcanizing agent and accelerators were added first, and the prepared masterbatch was further mixed with the rubber compound for only 2 minutes to avoid structural rupture and breakage of the GO particles. The final compound was cured and molded at 150 °C under a pressure of 10 MPa, then cooled for 5 minutes under a pressure of 5 MPa. The curing conditions were selected based on rheometric assessment. NR samples with 0, 0.4, 0.8 and 1.6 wt% GO were prepared and named G0, G0.4, G0.8 and G1.6, respectively.| Sample | Raw NR | Masterbatch | ZnO | SA | S | CZ | MBT |
|---|---|---|---|---|---|---|---|
| a All quantities are in parts per hundred (pph). | |||||||
| G0 | 100 | 0 | 5 | 2 | 2.5 | 0.9 | 0.12 |
| G0.4 | 96.3 | 3.7 | 5 | 2 | 2.5 | 0.9 | 0.12 |
| G0.8 | 92.6 | 7.4 | 5 | 2 | 2.5 | 0.9 | 0.12 |
| G1.6 | 85.2 | 14.8 | 5 | 2 | 2.5 | 0.9 | 0.12 |
Characterization
Fourier transform infrared (FTIR) spectroscopy of the samples was performed to evaluate the extent of oxidation and reduction. Samples were prepared as thin films and studied using a Nicolet-IR100 spectrometer. The NR–rGO samples were first frozen in liquid nitrogen and powdered; then mixed with KBR to obtain test samples. The spectrum was obtained from 400 to 4000 cm−1 and with a resolution of 4 cm−1.AFM analysis was performed using a DS-95-200E system in tapping mode. A dispersion of GO with a concentration of 0.02 mg ml−1 was prepared and deposited on a mica substrate as the test specimen.
To study the emulsion microstructure, optical micrographs of the Pickering emulsions were put on a transparent glass slide and observed using a Leica DMR microscope.
The fractured surface of the NR–rGO samples and GO sheets on silicon wafer were examined by scanning electron microscopy (SEM) using a Seron AIS2100 with a 15 kV accelerating voltage. The samples were coated with a thin layer of gold to prevent electron charging.
The layered structure of the GO and NR–rGO samples was examined by X-ray diffraction (XRD) using an EQuinox 3000 (Inel). Measurements were performed under standard laboratory conditions, using Cu Kα radiation (X-ray wavelength of 1.5406 Å).
To calculate the surface tension of the prepared GO, contact angle measurements were employed. A GO film was prepared through vacuum filtration and liquid droplets were placed on the film by a microsyringe. The images were instantly taken by a digital camera and the procedure was repeated 3 times for each liquid.
To evaluate the mechanical properties of the samples, uniaxial tensile tests were performed using a universal tensile machine under standard laboratory conditions. The samples were cut into dumbbell-shaped 10 mm × 2.5 mm × 1.5 mm pieces. The tests were all conducted at a constant crosshead speed of 500 mm min−1 and five specimens were tested for each composite sample.
Thermogravimetric analysis (TGA) was carried out using a TGA Q50 instrument under a nitrogen atmosphere, to determine the rGO concentration. A 4 mg sample was heated from 50 to 600 °C at a heating rate of 20 °C min−1.
Results and discussion
Self-assembly and dispersion of GO at oil–water interfaces
GO is known as a 2D Janus nanoparticle that is composed of distinguished hydrophilic and hydrophobic regions resulting in an amphiphilic nature, which makes it capable of stabilizing oil–water emulsions (Fig. S1 and Table S1, ESI†).21,35–37 It is observed that GO monolayers tend to self-assemble at air–water interfaces.38 Besides migration to the free surface of water, GO sheets are able to act as stabilizing agents in the production of air–water foams and water–air powders.38 This phenomenon is very similar to the oil–water emulsification process. It has also been shown that the stabilization by GO is not limited to liquid–liquid emulsions. Huang et al.33 has reported the stabilization of graphene and CNTs in aqueous media using GO as a dispersing agent.Commonly, GO is prepared by chemical oxidation/exfoliation of pristine graphite. Treating natural graphite in a mixture of H2SO4 and KMnO4 according to the Hummers method, or other chemical oxidation methods, leads to the formation of oxygenated functional groups such as carboxyls on the edges and hydroxyls, epoxides and ketones in the inner zone of the graphitic layers. As reported,31,33 the main functional groups which determine the hydrophilic behavior of GO are carboxyl groups, which acquire a negative electrostatic charge through reversible proton-exchange cycles in aqueous media.31 The hydrophobic character of GO may come from the graphitic domains surrounded by oxygenated groups, as shown in Scheme 1.31 Analogous to the pristine graphite, these domains are able to interact with hydrophobic media by van der Waals forces or π–π bond interactions through conjugated bonds. Better results are expected with aromatic liquids such as toluene and xylene due to the presence of the aromatic ring. The hydrophobic/hydrophilic ratio of GO is an important parameter which is tunable during GO synthesis.33,38 Therefore, selecting an appropriate synthesis procedure to obtain the desirable structural characteristics is of prime importance.
 | ||
| Scheme 1 Adsorption of GO layers to the surface of oil droplets (left); hydrophobic and hydrophilic zones on the GO surface (right). | ||
Several parameters drastically affect the final properties of the Pickering emulsion obtained with GO particles.33,39,40 Emulsion composition and the ratio of oil-to-water phases determine the emulsion type being o/w or w/o systems, also known as Pickering and inverse Pickering emulsions.41 The concentration of the GO stabilizing particles affects the size distribution of the emulsified droplets. The procedure and sequence of adding GO stabilizing particles to the oil–water mixture makes a considerable difference in the state of the final emulsion.36,41 The initial amount of energy given to the biphasic system is another factor that determines the droplets’ dimensions.36,41 To trigger an emulsification process, some initial energy is required to break the minor phase into the micro-droplets, which creates the oil–water surface area for stabilizing particles that move to the interface. The tunable amphiphilicity of GO introduces new variables such as pH of the medium, which substantially affect the degree of emulsification and emulsion properties. As reported,31,33,38 amphiphilicity and emulsification capability of GO is strongly dependent on the dimensional characteristics of the layers (Fig. S2 and S3, ESI†).31,42
Fig. 1 shows the effect of GO concentration on the droplet sizes. A transition from wholly merged oil droplets to the emulsified droplets is observed. Based on the optical microscopy images, at low GO concentrations, large droplets of the oil phase are formed in the aqueous phase that coalesce together soon after. It is because of the inequality between the created surface area of the droplets (that is inversely related to the initial given energy) and the available surface area of the GO sheets.41,43 In low GO concentrations, there are not enough sheets to cover all of the newly created oil–water interfacial area, thus the droplets tend to coalesce and form large droplets with a broad size distribution. As the concentration of GO increases, the droplets become surrounded by GO particles more and more. Consequently, the mean size of the droplets decreases and a narrower size distribution is obtained as the coalescence is avoided.41,43
 | ||
| Fig. 1 Effect of GO concentration on the size of droplets; (a)–(c) optical images at 0.18, 0.75 and 1.2 mg ml−1 GO concentration and (d) digital photograph of Pickering emulsions with 0.18–1.2 mg ml−1 GO concentration. | ||
Nevertheless, an excess amount of GO can negatively affect the homogenous dispersion, as there would not be enough interfacial area for all particles to migrate to. Subsequently, the excess particles remain in the aqueous phase, which can form aggregates as a result of restacking during the drying step, leading to partial flocculation in the masterbatch. Therefore, selecting the appropriate concentration of GO is important to prevent morphological heterogeneity. Moreover, the concentration of polymer (NR) in the oil phase (toluene) is of great significance, as this extremely affects the formation of emulsion droplets (Fig. S4, ESI†). For the 40:60 proportion of oil–water phases and a 20 mg ml−1 concentration of NR in toluene, it was found that roughly 12 wt% of GO (relative to the dissolved NR) is appropriate. It has also been observed that gradual addition of GO has an impact on Pickering emulsion formation, while the sudden addition of the total amount of GO results in a different emulsion state. This can be attributed to the change in the initial oil–water phase ratio that affects the final Pickering emulsion properties.
Immediately after churning, the two-phase liquid mixture turns into a single phase paste. The transition is due to the remarkable enhancement in the interfacial area between oil and water, associated with the formation of droplets and following the adsorption of particles. The entrapment and subsequent stabilization of air droplets is another effective factor. Fig. 2 shows the optical microscopy image of the prepared Pickering emulsion of NR solution after a month. Considerable stability of Pickering emulsions during long periods can be regarded as advantageous as it preserves the uniform dispersion of the GO layers, unlike that of the latex method. It is worthwhile mentioning that selection of the oily solvent is not restricted to toluene and there are other appropriate solvents that can be utilized (Table S2, ESI†).
 | ||
| Fig. 2 Final masterbatch; (a) optical image after a month, (b) paste-like Pickering emulsion and (c) after microwave drying. | ||
The distribution of nanoparticles through a polymeric phase is the first step toward fabricating a well-dispersed composite. The separation of continuous liquid phases for preparing the final masterbatch is the next challenge, which can be accompanied by partial flocculation of particles if performed inappropriately. Therefore, the design of a suitable drying procedure to vaporize aqueous and oily phases without aggregation of particles is extremely important. To show the effect of the drying procedure, identical samples of the prepared Pickering emulsion have been dried by two different approaches as shown in Scheme 2.
 | ||
| Scheme 2 Schematic of different drying approaches and subsequent masterbatch dispersion. | ||
Fig. 3 shows SEM images of the dried samples. Obvious phase separation and a layered structure of graphitic sheets are observed for the sample, which was dried in the oven. This can be explained as the result of the sequential evaporation of water and toluene that leads to separate precipitation stages of the polymeric and reinforcing phases.
 | ||
| Fig. 3 SEM images of masterbatch; (a) microwave and (b) oven-dried. | ||
In this case, toluene vaporizes completely before water does due to comparatively smaller vapour pressure and amount of the oil phase. Then, in the absence of an oil–water interface, GO layers freely transfer back to the aqueous phase. Also, in the absence of the oil phase, polymer chains, which are insoluble in water, separate and precipitate, while most of the GO layers are still dispersed in the aqueous phase. This leads to a noticeable phase separation in the final masterbatch.
On the other hand, no considerable aggregation of GO layers was detected throughout the masterbatch which was dried by microwave radiation, as shown in Fig. 3.
Microwave radiation initiates vaporization of water before toluene, preventing instant segregation of the polymeric phase. Water molecules evaporate due to the vibration of O–H bonds, but toluene experiences no vibrational movement under the radiation. So, evaporation of the whole oil phase is retarded, which leads to simultaneous precipitation of polymeric chains and GO layers.
Therefore, as the first and most essential consequence of microwave drying, the homogenous dispersion of layers throughout the mixture is preserved, due to decreasing the possibility of successful sedimentation of the polymer and particles. For a further study of dispersion, XRD patterns of GO and microwave dried samples are presented and compared in Fig. 4. A sharp peak at 2θ = 11° is observed which corresponds to a 0.8 nm interlayer distance for GO. However, the NR–rGO sample does not give rise to a distinctive peak, confirming good dispersion of layers throughout the polymeric matrix, which is in agreement with the observations from the SEM images.
 | ||
| Fig. 4 XRD patterns of GO and NR–rGO samples. | ||
Moreover, partial reduction of oxygen-rich groups on a GO surface by microwave radiation has also been reported.39,44,45 Fig. 5 portrays the normalized FTIR spectra of the synthesized GO and NR masterbatch, dried by microwave radiation. The presence of different oxygen-containing groups on the GO surface was confirmed by peaks at 3438, 1712, 1230 and 1054 cm−1, which correspond to the stretching vibrations of O–H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, C–OH and C–O bonds, respectively.46 On the other hand, the partial reduction of GO to rGO during microwave drying is conclusive, based on the significant attenuation of O–H bond peak intensity at 3438 cm−1. However, the CO bond peak at 1712 cm−1 and the C–O bond peak at 1054 cm−1 remain almost unchanged, which indicate the partial presence of oxygen-containing groups after reduction. So, through microwave drying, the graphitic structure of the layers is simultaneously retrieved without the need for using any chemical reducing agents such as hydrazine.
O, C–OH and C–O bonds, respectively.46 On the other hand, the partial reduction of GO to rGO during microwave drying is conclusive, based on the significant attenuation of O–H bond peak intensity at 3438 cm−1. However, the CO bond peak at 1712 cm−1 and the C–O bond peak at 1054 cm−1 remain almost unchanged, which indicate the partial presence of oxygen-containing groups after reduction. So, through microwave drying, the graphitic structure of the layers is simultaneously retrieved without the need for using any chemical reducing agents such as hydrazine.
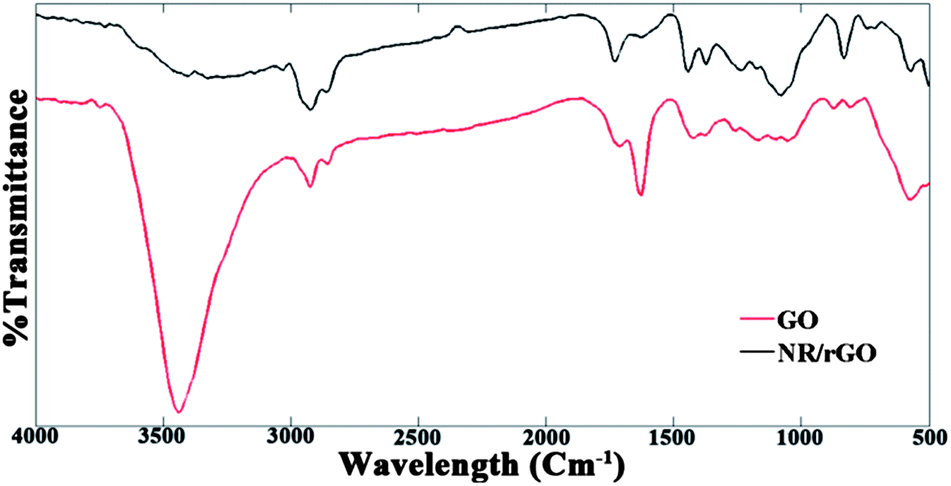 | ||
| Fig. 5 FTIR spectra of GO and NR–rGO samples. | ||
Besides partially restoring the electrical conductivity of particles, reduction of polar groups can be advantageous for improving the bonding of particles with a NR matrix, with regards to the chemical structure of the chains. In the latex method, chemical reduction of GO can diminish the electrostatic stability of layers in an aqueous medium,47 and consequently the homogeneity of the polymer–graphene mixture. So, chemical reduction in the latex method can be accompanied by a negative effect on the degree of dispersion, regardless of other disadvantageous facets such as toxicity of the chemicals.
Fig. 6 shows the TGA and DTGA graphs of the NR–rGO sample. An 11% reduction in total weight occurs between 120 and 350 °C, which is attributed to the degradation of rGO.48,49 A sharp decline in total weight is initiated at 350 °C and a single peak in the DTGA graph at 380 °C indicates a weight reduction due to elastomer degradation.50 Based on Fig. 6, the filler content of the masterbatch is then determined to be 11 wt%. This value is almost consistent with the relative proportions of GO and NR (approximately 12 wt%) employed in the preparation of the Pickering emulsion.
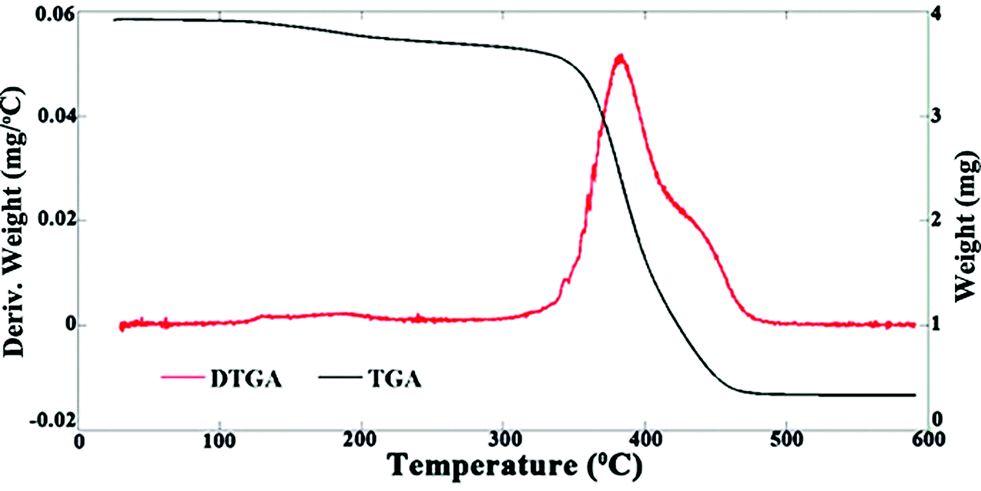 | ||
| Fig. 6 TGA (left axis) and DTGA (right axis) graphs of the masterbatch. | ||
Mechanical properties of the NR–rGO composite
Several factors are known to contribute in mechanical reinforcement of polymeric composites. Besides the hydrodynamic effect due to the inclusion of solid particles,51 formation of a strong interface between the polymeric matrix and particles plays an important role in mechanical reinforcement. It can be concluded that the mechanical reinforcement efficiency is in direct correlation with the established interfacial area and the strength of it, which is dependent on the dispersion effectiveness per se. Appropriate dispersion promises a large interfacial area which efficiently transfers the exerted load to the particles with a lower stress concentration.2 Moreover, it is essential to preserve the perfect pristine structure of sheets through the dispersion procedure. Fabrication methods such as severe oxidizing conditions or intensive ultrasonic treatment cause structural defects. Two types of structural defects are possible during the dispersion stage, which lower the reinforcing efficiency of GO/rGO composites.52First, there are local defects at the oxidized zones as a result of chemical exfoliation and reduction. Second are the structural defects caused by tearing and rupture of the GO layers, leading to a significant decrease in the lateral dimension of particles. Compounding by mechanical mixers such as a two-roll mill or internal mixer for long periods destroys the GO structure as well.53 So, improving the dispersion of layers through the masterbatch preparation and reducing the need for mechanical mixing is an appropriate strategy to preserve the structure of graphene.
In this case, fabricating composites based on the self-assembly of particles is advantageous, considering the minimal sonication and mechanical mixing that delivers a good dispersion with the least structural damage to GO/rGO.
Stress–strain curves of NR–rGO with three different amounts of rGO are given in Fig. 7. Tensile properties are presented in Table 2. Obviously, the inclusion of rGO particles has increased the mechanical characteristics, especially at high elongation ratios.54,55 Table 2 shows that addition of only 0.8 wt% rGO results in a 35% increase in stress at 300% elongation and 39% improvement in the ultimate tensile strength of the composite. This improvement may be explained by several reasons as follows: outstanding intrinsic mechanical characteristics of the graphene monolayer, higher specific surface area compared to 1D CNTs and microspherical CB and appropriate interactions with NR chains, which can be analogous to additional entanglements or physical crosslinking, are effective parameters. A flexible structure combined with the wrinkled topology of rGO layers results in a surface roughness on the nanoscale, which can improve adhesion and interaction of the layers with polymeric chains through a mechanical interlocking mechanism.56 Also, it is demonstrated that strain-induced crystallization as a substantial feature in mechanical reinforcement of NR specifically at high elongation ratios is significantly intensified in the presence of rGO layers.57
 | ||
| Fig. 7 Stress–strain curves of NR–rGO nanocomposites. | ||
| Sample | Tensile strength (MPa) | Elongation at break (%) | Stress at strain 100% (MPa) | Stress at strain 300% (MPa) |
|---|---|---|---|---|
| G0 | 7.52 | 602 | 0.56 | 1.43 |
| G0.4 | 8.58 | 607 | 0.58 | 1.59 |
| G0.8 | 10.47 | 598 | 0.63 | 1.99 |
| G1.6 | 7.88 | 482 | 0.69 | 2.98 |
Neglecting the small increase for the G0.4 sample which can be assigned to instrumental error, the elongation at break for the G0.4 and G0.8 samples is nearly identical to the neat NR. This can be correlated to the uniform dispersion of rGO layers and the formation of an appropriate interfacial area between particles and chains that reduces the possibility of particles’ aggregation. The flexible structure and wrinkled topology of the GO monolayers is another effective parameter for reducing stress concentration over contact sites, which allows polymeric chains to possess local motions.
For further investigation, Mooney–Rivilin curves were plotted based on eqn (1):58
| σ = 2(C1 + C2λ−1)(λ − λ−2) | (1) |
 | ||
| Fig. 8 Mooney–Rivilin curves of NR–rGO nanocomposites. | ||
C2, which is the slope of the curve, is directly correlated to the concentration of physical or unstable bonds and networks. For an elastomer with purely entropic elastic deformations known as a Gaussian network, C2 equals zero. The presence of active fillers such as rGO causes deviation from entropic elastic networks. Mutual interactions of particles form aggregated networks that are mechanically destroyed while the chemical network remains. Mechanical breakdown of the filler network and slippage at the interface is known as “stress softening”, “Mullins effect” or “Payne effect”.59 So, C2 is directly related to stress softening. As seen in Fig. 8, the curves at different rGO loadings are almost parallel at low elongation ratios, which imply that C2 is nearly independent of rGO concentration. This alludes to the appropriate dispersion and lack of aggregation of particles, even at high concentrations of filler.
The enhancement in reduced stress at high elongation ratios (λ−1 < 0.4) is assigned to the strain-induced crystallization. It shows that the presence of rGO layers intensifies strain-induced crystallization as the reduced stress increases noticeably compared with the neat NR. Also, with increasing addition of rGO, the point at which crystallization is initiated shifts to lower elongation ratios. This also indicates that rGO layers facilitate alignment and ordering of NR chains. rGO sheets tend to align in the direction of extensional force. Supposedly, this orientation imposes steric restrictions, which can lower the total entropy of the NR chains. So, orientation and ordering of chains can be facilitated in the presence of rGO layers.
Moreover, based on the stress–strain curves, it is conclusive that mechanical reinforcement by rGO is more significant and efficient at higher elongation ratios. Hypothetically, this can be assigned to the flexible structure of the graphene layers. With an atomic scale thickness and micron-sized 2D geometry, wrinkling and crumpling are likely for graphene nanolayers.56,60,61 As shown in Scheme 3, it can be speculated that most of the rGO layers are dispersed in a random orientation in a folded state and that extension of the composite, specifically at high levels, leads to gradual unfolding and arrangement of layers in the direction of elongation. The first possible consequence of this phenomenon is reduction of the stress concentration at the polymer–particle interfacial area. Secondly, with the orientation of unfolded sheets along the elongation direction, a greater fraction of layers are stretched under an in-plane loading state, which can significantly improve the load-bearing ability of the composite. On the contrary, for rigid particles with low deformability, drastic local stress intensification and interfacial cleavage is expected.
 | ||
| Scheme 3 Orientation and unfolding of crumpled layers due to extension. | ||
So, as a speculation, uniform dispersion of flexible but stiff rGO layers, which are capable of stretching and unfolding, can considerably improve the mechanical performance of polymers, without sacrificing inherent properties such as high elongation at break or damping capability (Fig. S5 and S6, ESI†).
To conclude, the main difference between the proposed procedure and the previously reported latex method11,14 lies in the driving force for the dispersion of nanoparticles throughout the polymeric phase. The dispersion of components in the latex method is due to the mechanical work performed on the system during sonication, while a Pickering emulsion results in dispersion due to the thermodynamically favorable self-assembly of GO particles. In the latex method, it takes a long sonication time to ensure complete homogenization. Besides, the mixture homogeneity reduces over time as there is not a strong interactional tendency between the GO particles and polymeric chains. The separation of water is another challenge with this method. Both evaporation and coagulation procedures are associated with the partial phase separation of particles, which can be the result of an unequal rate of sedimentation of polymeric chains and GO layers. Moreover, chemical reduction of GO by hydrazine, during separation of the aqueous medium, results in probable aggregation of rGO particles, which is detrimental to the dispersion and final properties of the composite. In the Pickering emulsion method, after providing the initial energy to create the interfacial area, GO layers are adsorbed by the polymeric phase instantly, therefore, the necessity for a sonication treatment is eliminated, which reduces the possibility of structural defects. Moreover, the prepared Pickering emulsion is stable even for months with no separation of phases due to the high energy holding the adsorbed GO particles around the droplets.41 Homogenous dispersion of layers throughout the polymer chains is preserved using the proposed microwave-drying procedure, which is also accompanied by partial reduction of GO. Moreover, unlike the latex method, which is restricted to a definite number of polymers, it is plausible to employ the Pickering emulsion method to fabricate composites of a wide range of polymers. As explained, the basic driving force beyond self-assembly and dispersion of GO layers around droplets of the polymer solution is the reduction of total free energy, due to replacement of the high energy oil–water surface area. So, it can be speculated that the interfacial tension of the oil phase (although it is slightly affected by dissolving a polymer) is the determining factor in the adsorption of layers, not the type of polymeric chains which are dissolved in the oily solvent. Therefore, it is suggested that a wide range of polymers, i.e. polyethylene, polypropylene, polystyrene and polyvinyl chloride with solubility parameters close to that of the employed oily solvent (toluene in this work) can be plausibly reinforced by rGO through the Pickering emulsion method in an efficient manner.
Conclusions
Graphene with an extraordinary specific surface area and superb intrinsic mechanical characteristics is an appropriate reinforcing particle for polymers. The challenging step is to disperse nanolayers uniformly in-between polymeric chains, without structural damage to the particles. Due to their amphiphilic nature, GO Janus layers tend to self-assemble at oil–water interfaces. Our results show that this intrinsic feature serves as an ideal tool to disperse GO in highly viscoelastic polymers spontaneously. A Pickering emulsion of a NR oily solution and water was prepared without sonication and dried by microwave radiation to keep the particles well dispersed, and a masterbatch of NR with 11 wt% rGO was produced. SEM, XRD and FTIR results show a homogenous dispersion of partially reduced GO layers in the prepared masterbatch.Notes and references
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- M. A. Rafiee, J. Rafiee, Z. Wang, H. Song, Z.-Z. Yu and N. Koratkar, ACS Nano, 2009, 3, 3884–3890 CrossRef CAS PubMed.
- S. Stankovich, D. A. Dikin, G. H. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282–286 CrossRef CAS PubMed.
- R. Verdejo, M. M. Bernal, L. J. Romasanta and M. A. Lopez-Manchado, J. Mater. Chem., 2011, 21, 3301–3310 RSC.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- N. Martin, M. A. Herranz and L. Rodriguez, Chem. Commun., 2013, 49, 3721–3735 RSC.
- J. Che, L. Shen and Y. Xiao, J. Mater. Chem., 2010, 20, 1722–1727 RSC.
- X. Huang, X. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- B. Chen, N. Ma, X. Bai, H. Zhang and Y. Zhang, RSC Adv., 2012, 2, 4683–4689 RSC.
- Z. Tang, Q. Wei, T. Lin, B. Guo and D. Jia, RSC Adv., 2013, 3, 17057–17064 RSC.
- Y. Zhan, J. Wu, H. Xia, N. Yan, G. Fei and G. Yuan, Macromol. Mater. Eng., 2011, 296, 590–602 CrossRef CAS.
- Y. Zhan, M. Lavorgna, G. Buonocore and H. Xia, J. Mater. Chem., 2012, 22, 10464–10468 RSC.
- P. Xie, X. Ge, B. Fang, Z. Li, Y. Liang and C. Yang, Colloid Polym. Sci., 2013, 291, 1631–1639 CAS.
- J. S. Kim, J. H. Yun, I. Kim and S. E. Shim, J. Ind. Eng. Chem., 2011, 17, 325–330 CrossRef CAS PubMed.
- J. Wang, H. Jia, Y. Tang, D. Ji, Y. Sun, X. Gong and L. Ding, J. Mater. Sci., 2013, 48, 1571–1577 CrossRef CAS.
- J. R. Potts, O. Shankar, L. Du and R. S. Ruoff, Macromolecules, 2012, 45, 6045–6055 CrossRef CAS.
- C. Li, C. Feng, Z. Peng, W. Gong and L. Kong, Polym. Compos., 2013, 34, 88–95 CrossRef.
- M. M. Gudarzi and F. Sharif, Soft Matter, 2011, 7, 3432–3440 RSC.
- A. Schrade, K. Landfester and U. Ziener, Chem. Soc. Rev., 2013, 42, 6823–6839 RSC.
- X. Song, Y. Yang, J. Liu and H. Zhao, Langmuir, 2011, 27, 1186–1191 CrossRef CAS PubMed.
- W. L. Zhang, Y. D. Liu, H. Choi and Y. Seo, RSC Adv., 2013, 3, 11723–11731 RSC.
- X. Hu, Z. Xu, Z. Liu and C. Gao, Sci. Rep., 2013, 3 Search PubMed.
- L. Kou and C. Gao, Nanoscale, 2013, 5, 4370–4378 RSC.
- D. Wu, F. Zhang, H. Liang and X. Feng, Chem. Soc. Rev., 2012, 41, 6160–6177 RSC.
- Y. Xu and G. Shi, J. Mater. Chem., 2011, 21, 3311–3323 RSC.
- D. Yu and L. Dai, J. Phys. Chem. Lett., 2010, 1, 467–470 CrossRef CAS.
- K. W. Putz, O. C. Compton, M. J. Palmeri, S. T. Nguyen and L. C. Brinson, Adv. Funct. Mater., 2010, 20, 3322–3329 CrossRef CAS.
- H. Bai, C. Li, X. Wang and G. Shi, Chem. Commun., 2010, 46, 2376–2378 RSC.
- J. Thieme, S. Abend and G. Lagaly, Colloid Polym. Sci., 1999, 277, 257–260 CAS.
- H. Chen, Z. Song, X. Zhao, X. Li and H. Lin, RSC Adv., 2013, 3, 2971–2978 RSC.
- J. Kim, L. J. Cote and J. Huang, Acc. Chem. Res., 2012, 45, 1356–1364 CrossRef CAS PubMed.
- Y. He, F. Wu, X. Sun, R. Li, Y. Guo, C. Li, L. Zhang, F. Xing, W. Wang and J. Gao, ACS Appl. Mater. Interfaces, 2013, 5, 4843–4855 CAS.
- L. J. Cote, J. Kim, V. C. Tung, J. Luo, F. Kim and J. Huang, Pure Appl. Chem., 2011, 83, 95–110 CrossRef CAS.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef.
- L. Hu, M. Chen, X. Fang and L. Wu, Chem. Soc. Rev., 2012, 41, 1350–1362 RSC.
- A. F. Mejia, A. Diaz, S. Pullela, Y.-W. Chang, M. Simonetty, C. Carpenter, J. D. Batteas, M. S. Mannan, A. Clearfield and Z. Cheng, Soft Matter, 2012, 8, 10245–10253 RSC.
- K. Bramhaiah and N. S. John, RSC Adv., 2013, 3, 7765–7773 RSC.
- J. Kim, L. J. Cote, F. Kim, W. Yuan, K. R. Shull and J. Huang, J. Am. Chem. Soc., 2010, 132, 8180–8186 CrossRef CAS PubMed.
- A. Kumar, B. J. Park, F. Tu and D. Lee, Soft Matter, 2013, 9, 6604–6617 RSC.
- B. P. Binks, R. Murakami, S. P. Armes and S. Fujii, Langmuir, 2006, 22, 2050–2057 CrossRef CAS PubMed.
- S. Melle, M. Lask and G. G. Fuller, Langmuir, 2005, 21, 2158–2162 CrossRef CAS PubMed.
- L. J. Cote, J. Kim, Z. Zhang, C. Sun and J. Huang, Soft Matter, 2010, 6, 6096–6101 RSC.
- S. Tarimala and L. L. Dai, Langmuir, 2004, 20, 3492–3494 CrossRef CAS.
- A. V. Murugan, T. Muraliganth and A. Manthiram, Chem. Mater., 2009, 21, 5004–5006 CrossRef CAS.
- Z. Li, Y. Yao, Z. Lin, K.-S. Moon, W. Lin and C. Wong, J. Mater. Chem., 2010, 20, 4781–4783 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- S. Bai, X. Shen, G. Zhu, A. Yuan, J. Zhang, Z. Ji and D. Qiu, Carbon, 2013, 60, 157–168 CrossRef CAS PubMed.
- Y. Zhang, J. Ge, L. Wang, D. Wang, F. Ding, X. Tao and W. Chen, Sci. Rep., 2013 DOI:10.1038/srep02771.
- N. Yooprasert, T. Pongprayoon, P. Suwanmala, K. Hemvichian and G. Tumcharern, Chem. Eng. J., 2010, 156, 193–199 CrossRef CAS PubMed.
- L. Gong, I. A. Kinloch, R. J. Young, I. Riaz, R. Jalil and K. S. Novoselov, Adv. Mater., 2010, 22, 2694–2697 CrossRef CAS PubMed.
- J. W. Suk, R. D. Piner, J. An and R. S. Ruoff, ACS Nano, 2010, 4, 6557–6564 CrossRef CAS PubMed.
- N. Jing, Q. Xue, C. Ling, M. Shan, T. Zhang, X. Zhou and Z. Jiao, RSC Adv., 2012, 2, 9124–9129 RSC.
- J. R. Potts, D. R. Dreyer, C. W. Bielawski and R. S. Ruoff, Polymer, 2011, 52, 5–25 CrossRef CAS PubMed.
- S. Thomas, Advances in Elastomers II: Composites and Nanocomposites, 2013, p. 1 Search PubMed.
- X. Ma, M. R. Zachariah and C. D. Zangmeister, Nano Lett., 2012, 12, 486–489 CrossRef CAS PubMed.
- B. Ozbas, S. Toki, B. S. Hsiao, B. Chu, R. A. Register, I. A. Aksay, R. K. Prud'homme and D. H. Adamson, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 718–723 CrossRef CAS.
- U. Eisele, Introduction to polymer physics, Springer-Verlag, 1990 Search PubMed.
- A. R. Payne, J. Appl. Polym. Sci., 1962, 6, 57–63 CrossRef CAS.
- J. Zang, S. Ryu, N. Pugno, Q. Wang, Q. Tu, M. J. Buehler and X. Zhao, Nat. Mater., 2013, 12, 321–325 CrossRef CAS PubMed.
- X. Ma, M. R. Zachariah and C. D. Zangmeister, J. Phys. Chem. C, 2013, 117, 3185–3191 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ra46342g |
| This journal is © The Royal Society of Chemistry 2014 |