
Unraveling the interplay of different contributions to the stability of the quinhydrone dimer
Abstract
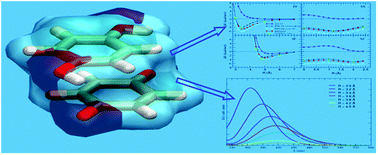
Aim of this paper is to present a computational revisitation of the main structural and spectroscopic features of quinhydrone, a prototype of complexes built on noncovalent interactions, with a view to proposing an accurate yet computationally convenient approach to the characterization of such kind of complexes. Several methods are compared in terms of energy profiles along selected coordinates, which involve the relative distance and/or orientation of the two aromatic rings. MP2 and DFT calculations agree in indicating that H-bonding and dispersion forces play a more important role than charge transfer in stabilizing quinhydrone. Distance- and orientation-dependent overlap of π clouds was found by TD-DFT calculations to be a major determinant of quinhydrone visible absorption and color.
 Please wait while we load your content...
Please wait while we load your content...

