
PhIO promoted synthesis of nitrile imines and nitrile oxides within a micellar core in aqueous media: a regiocontrolled approach to synthesizing densely functionalized pyrazole and isoxazoline derivatives†
Abstract
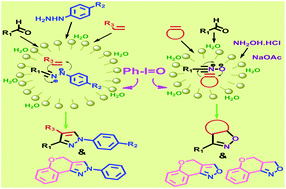
A highly efficient and general protocol has been developed for the facile synthesis of highly diversified 1,3,5-trisubstituted pyrazoles and 3,5-disubstituted 2-isoxazolines through one-pot tandem intermolecular, as well as intramolecular, dipolar [3 + 2] cycloaddition reactions. Chemoselective oxidation of aldohydrazone to nitrile imine and aldoximes to nitrile oxides by iodosobenzene in neutral aqueous media was performed. The scope of the reactions in regiocontrolled dipolar cycloaddition with olefins in the presence of PhIO toward highly substituted pyrazole and isoxazoline derivatives has been demonstrated. SDS converted the initially floating reaction mass and the organo-oxidant PhIO into a homogeneous mixture, which on stirring became a turbid emulsion. The micellar arrangement was confirmed by dynamic light scattering and optical microscopy. The new, green and metal free synthetic method enabled the execution of regiocontrolled tandem cycloaddition oxidation sequences leading to a library of pyrazole and isoxazoline derivatives.
 Please wait while we load your content...
Please wait while we load your content...

