
High-efficiency water oxidation and energy storage utilizing various reversible redox mediators under visible light over surface-modified WO3†
Abstract
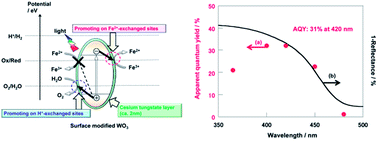
Tungsten trioxide (WO3) powder, treated with various metal salt solutions, was used for the photocatalytic oxidation of water into O2; the reaction was accompanied by the reduction of various redox oxidants. The photocatalytic activity of WO3 was remarkably improved by thermal treatment with alkali metal and silver salt aqueous solutions. Cs-treated WO3 showed the highest activity. The WO3 particles were covered with a very thin layer of a cesium tungstate species, and the ion-exchangeable sites on the WO3 surface were formed by Cs-treatment. The activity of Cs-treated WO3 was further improved by ion exchange of H+ and Fe2+ ions and was 24 times higher than the activity of WO3 without treatment. All methods of O2 evolution using IO3− and VO2+ on WO3 were also significantly improved with Cs-treatment, which suppressed the reverse reaction of the oxidation of the various redox reductants. The optimized WO3 in Fe(ClO4)3 aqueous solution had a high quantum yield (31% at 420 nm) and solar-to-chemical energy conversion efficiency (0.38%).
 Please wait while we load your content...
Please wait while we load your content...

