
Self-regenerative adsorbent based on the cross-linking chitosan for adsorbing and mineralizing azo dye†
Abstract
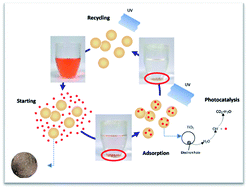
In this work, we synthesized self-regenerative chitosan (SRCS) by combining the photo-catalyst TiO2 and cross-linking chitosan (CS). The novel SRCS hybrids were then used as adsorbents to remove methyl orange (MO) and they demonstrated excellent adsorption capacity (∼799.2 mg g−1) and significant photocatalytic self-regenerative properties. The pseudo-first-order (PFO), pseudo-second-order (PSO) and Weber–Morris kinetics models were applied to fit the experimental data obtained from batch experiments. The PSO kinetic model was more appropriate for describing the adsorption of MO onto the SRCS. Interestingly, the SRCS adsorbent was successfully regenerated by UV photocatalysis after adsorption. More importantly, the adsorption effectiveness of the SRCS remained constant through eight regeneration cycles. This study provides a green method of removing organic pollutants that combines adsorption enrichment with photocatalytic degradation.
 Please wait while we load your content...
Please wait while we load your content...

