Nitrogen- and oxygen-containing activated carbon nanotubes with improved capacitive properties†
Zhenfang Zhoua,
Zhonghua Zhanga,
Hongrui Penga,
Yong Qinb,
Guicun Li*a and
Kezheng Chen*a
aLaboratory of Functional and Biological Nanomaterials, College of Materials Science and Engineering, Qingdao University of Science and Technology, Qingdao 266042, China. E-mail: guicunli@qust.edu.cn; kchen@qust.edu.cn
bState Key Laboratory of Coal Conversion, Institute of Coal Chemistry, Chinese Academy of Sciences, Taiyuan, 030001, China
First published on 17th December 2013
Abstract
Nitrogen- and oxygen-containing activated carbon nanotubes have been successfully synthesized via a high temperature carbonization of polypyrrole (PPy) nanotubes followed by chemical activation. The first carbonization step ensures the formation of amorphous carbon nanotubes, which are more stable than PPy to preserve the nanotube morphology during the subsequent chemical activation process. The obtained activated carbon nanotubes with high nitrogen (19.8 wt%) and oxygen contents (11.1 wt%) show enlarged specific areas of 705.9 m2 g−1 compared to that of the pristine carbon nanotubes (212.4 m2 g−1). As expected, the activated carbon nanotubes exhibit enhanced capacitance properties, such as an enlarged specific capacity (384.9 F g−1 at 0.5 A g−1), excellent rate capability (201 F g−1 at 50 A g−1), and more stable cyclic stability (only 2.4% of specific capacitance loss after 500 cycles) due to its abundant micropores, high specific areas and abundant interfacial functional groups. This method is facile, low cost and enables easy production of large quantities, and can be expected to open up new opportunities in designing high-performance carbon electrode materials for supercapacitors.
Introduction
Supercapacitors, also known as electrochemical capacitors, have attracted tremendous attention due to unique dual advantages of both traditional dielectric capacitors and rechargeable batteries, which can transport high power within a very short period, store high energy, and exhibit closely infinite long cycle life.1–3 They are considered to be very promising energy storage devices in a wide range of applications, such as digital devices, electrical vehicles, regenerative energy, etc.1,4,5 In recent years, major progress has been made in the theoretical research and practical development of supercapacitors. Carbon-based materials, such as activated carbons,6–10 mesoporous carbons,11,12 carbon nanotubes,13–15 and graphene,16–18 have been intensively investigated as electrode materials for electrochemical double-layer capacitors (EDLC). It is reported that activated carbon materials have been successfully developed as electrodes for commercial supercapacitors because of their high specific surface area and low cost.6–9 They can deliver high specific capacitance at low discharging rate, their high rate capability is unsatisfactory mainly due to low electrode/electrolyte accessibility lead by intrinsically high fraction of microporosity.6,10 Carbon nanotubes have been considered as one of the most promising electrode materials for supercapacitors due to their well-defined porous nanostructure and high electronic conductivity.13–15 Unfortunately, their relative low specific surface area limits their EDLC capacitance.19To meet acceptable capacitance of carbon based electrode materials, great efforts have been focused on the design of nanoscale carbon materials with high active surface areas,20–23 incorporating pseudocapacitive materials,24,25 and doping heteroatoms such as nitrogen,26–30 sulfur31,32 and oxygen,33,34 etc. Among them, the introduction of nitrogen functional groups into carbon-based materials seems to be the most promising method for improving their electrochemical performances due to the enhanced surface activity, the inherent electrical properties and capacitive enhancement through Faradic pseudocapacitive reactions.28–33 Generally, two strategies for achieving nitrogen doped activated carbons are post-treatment of carbons with ammonia gas26,27 and using nitrogen containing precursor.28–30 The post-treatment process requires the high temperature and toxicity nitrogen containing atmosphere (e.g., NH3) and usually leads to a decrease in surface area and structural degradation.35,36 In addition, nitrogen-containing functional groups developed in such a way are unstable and the nitrogen is mainly distributed on their surface28,30 Direct synthesis from nitrogen-containing precursor can avoid these disadvantages. It has been established that nitrogen-containing carbon materials derived from different precursors (such as poly(o-phenylenediamine),35 PPy,37,38 polyaniline,39 etc.) are promising for application in lithium ion batteries and supercapacitors. For examples, Huang et al.37 have reported nitrogen doped porous carbon nanofiber derived from PPy nanofibers as anodes for lithium ion batteries with a super high capacity and rate capability. Lou et al.12 have investigated the improved electrochemical capacitance of PPy-derived microporous carbon nanospheres. Yu et al.38 have presented a high-capacity supercapacitor material based on the nitrogen-doped porous carbon nanofibers synthesized by carbonization of carbonaceous nanofibers coated with PPy, which exhibit a reversible specific capacitance of 202.0 F g−1 at the current density of 1.0 A g−1 in 6.0 mol L−1 aqueous KOH electrolyte. The introduction of oxygen surface groups into carbon-based materials, usually formed in carbonization of oxygen containing carbon source and/or in most of the activated carbons during the activation process, is also reported to be another promising approach to enhance the capacitive performances because their acidic character can introduce electron-acceptor properties into the carbon surfaces, which can give rise to pseudocapacitive reactions.33,40–44 Bandosz et al.33 have reported microporous activated carbon treated by urea or melamine to achieve the combined effect of nitrogen- and oxygen-containing functional groups. Ma et al.42 have showed that relative stable oxygen groups in carbon monolith can not only strengthen the wettability of the interface between electrode and electrolyte, but also introduce pseudocapacitive effects. Herein, we report a facile strategy to synthesize activated carbon nanotubes with high contents of nitrogen- and oxygen-containing functional groups via a carbonization and activation process using PPy nanotube as precursor. The synthesized nitrogen- and oxygen-containing activated carbon nanotubes exhibit excellent capacitive performance, particularly high rate capability and long cyclic stability.
Experimental
Synthesis of PPy-derived activated carbon nanotubes
PPy nanotubes were synthesized using a self-degraded template method as previous reported.45 In a typical procedure, 16.2 g of FeCl3 was dissolved in 2 L of 5 m mol L−1 sodium 4-[4′-(dimethylamino)phenyldiazo] phenylsulfonate aqueous solution. 3.5 mL of pyrrole monomer was added into the above solutions and the mixture was stirred at room temperature for 24 h. The formed PPy precipitates were filtrated and washed with NH4·OH solution to eliminate of Cl− ions, followed by filtrations with deionized water and ethanol several times until the filtrate was colorless and neutral. The PPy precursor was finally dried at 60 °C for 24 h. The first carbonization step was performed at 700 °C in N2 for 2 h to obtain nitrogen containing amorphous carbon nanotubes (CN). The amorphous carbon nanotubes were mixed with KOH at a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, and then heated to 700 °C in N2 for 1 h. After the furnace cooling down to room temperature, the obtained mixtures were washed with dilute HCl solution to remove inorganic salts and then washed with distilled water until neutral pH. The final products were obtained after drying at 120 °C overnight and labelled as ACN.
:2, and then heated to 700 °C in N2 for 1 h. After the furnace cooling down to room temperature, the obtained mixtures were washed with dilute HCl solution to remove inorganic salts and then washed with distilled water until neutral pH. The final products were obtained after drying at 120 °C overnight and labelled as ACN.
Characterization
The morphologies and sizes of the products were characterized using a field-emission scanning electron microscopy (FE-SEM, JSM 6700F) and a transmission electron microscopy (TEM, FEI Tecnai G20, USA). The X-ray photoelectron spectroscopy (XPS) analysis was performed on a Perkin-Elmer PHI 550 spectrometer with Al Kα (1486.6 eV) as the X-ray source. The curve fittings of N1s and O1s envelopes were performed with a mixed Gaussian (20%)–Lorentzian (80%) component profiles after subtraction of a Shirley background with using XPS peak processing software.46 The fitting was performed by fixing the peak maximum within ±0.1 eV for all spectra and applying a full width half-maximum of 1.4–1.6 eV. All peak positions were calibrated with the graphitic carbon peak at 284.6 eV, and the elemental compositions were determined from peak area ratios after correction for the sensitivity factor for each element. Nitrogen sorption isotherms of the samples were measured at 77 K with a micrometrics ASAP 2020 analyzer. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface area using adsorption data in a relative pressure range from 0.04 to 0.2. The pore size distribution was computed by the nonlocal density functional theory (NLDFT) method.Electrochemical test
The working electrodes were fabricated by mixing the products and polytetrafluoroethylene (PTFE) in the mass ratio 95:5. The mixtures were dissolved in ethanol and grinded adequately to form slurry. Then, the slurry was pasted onto a stainless steel electrode (0.5 cm × 0.5 cm) and dried at 80 °C for 24 h under vacuum. The mass loading of active materials in each electrode was calculated to be 1.5 mg cm−2 by weighting the carbon loaded electrode and stainless steel strip. The electrochemical properties of supercapacitor electrodes were measured in three-electrode systems, in which a platinum electrode, saturated calomel electrode (SCE), and 1 mol L−1 H2SO4 aqueous solution were used as counter electrode, reference electrode, and electrolyte, respectively. The capacitive performances of the samples were investigated by using cyclic voltammetry (CV), galvanostatic charge–discharge, and electrochemical impedance spectroscopy (EIS) techniques, which were performed using an Autolab PGSTAT302N electrochemical workstation. CV measurements were performed at a range of scan rate from 5 to 100 mV s−1. The specific capacitance of the electrode can be calculated according to the equation: C = ∫IdV/(νmΔV), where I is the response current density (A g−1), V is the potential (V), ΔV is the potential window, ν is the potential scan rate (mV s−1), and m is the active mass in the electrodes (g). EIS measurements were carried out applying a sine wave with the amplitude of 10.0 mV over the frequency range from 100 kHz to 10 mHz. The galvanostatic charge–discharge tests were performed at a range of current rate from 0.5 to 50 A g−1. On the basis of the galvanostatic charge–discharge curves in three-electrode system, the specific capacitances C (F g−1) of the electrode can be calculated according to the following equation: C = IΔt/(ΔVm), where I is the discharge current (A), Δt is the discharge time (s), ΔV is the potential change during the discharge process, and m is the active mass of electrode (g).
Results and discussion
SEM image of PPy precursor (Fig. S1a, ESI†) shows a uniform fibrillar morphology with average outer diameter in the range of 170–220 nm and length up to several micrometers, which is different from the granular morphology by conventional synthesis. TEM image (Fig. S1b and c, ESI†) reveals the hollow tubular morphology feature of the PPy precursor with close ends. PPy nanotubes were used as nitrogen-containing precursor for the synthesis of CN and ACN by a facile carbonization and activation process. Fig. 1 presents typical SEM, TEM and High-resolution TEM (HRTEM) image images of CN and ACN samples. As shown in Fig. 1a and b, it is clear that the CN sample preserves the tubular morphology of the PPy nanotubes after carbonizations at 700 °C. Lots of CN are fractured to form open ends as indicated by arrows (Fig. 1b) mainly due to the pyrolysis of PPy precursor. The outer diameters of CN are decreased to 140–180 nm. High-magnification TEM image in Fig. 1c shows a typical CN with rough walls and a open end. HRTEM image (Fig. 1d) reveals that CN has dense and amorphous carbon structures without observing any crystalline impurities and graphitic species. After activated at 700 °C by KOH, bundle-like ACN are formed (Fig. 1e and f). The tubular morphology is not destroyed during the activation process. The outer diameters of ACN are further decreased to 120–160 nm. High-magnification TEM image of ACN (Fig. 1g) shows that the walls become rougher than that of CN, indicating that KOH can penetrate into the amorphous CN and etch the amorphous carbon frameworks by chemical reactions. In addition, the ACN with open-ended tips can provide a buffering reservoir for electrolytes to minimize the diffusion distances to the interior surfaces of the pores, so both outer surfaces and inner surfaces of ACN are available for ion transport/charge storage. HRTEM image of ACN (Fig. 1h) presents some discrete graphitic layers on the wall of the carbon nanotubes with highly developed porous structures. | ||
| Fig. 1 SEM images of CN (a) and ACN (e) showing randomly fibrillar morphology. TEM images of CN (b and c) and ACN (f and g) showing tubular morphology. HRTEM images of CN (d) and ACN (h). | ||
N2 absorption–desorption isotherms and the derived pore-size distributions of CN and ACN are plotted in Fig. 2a and b, respectively. Both isotherms (Fig. 2a) can be classified as a type IV isotherm with mesoporous features. The strong N2 adsorption below the relative pressure of P/P0 = 0.1 for ACN is a feature of micropore filling. The gradually increasing in the middle-pressure region (P/P0 = 0.15–0.8) of the adsorption curve from both CN and ACN implies the presence of mesopores. While the abruptly increasing in the high-pressure region (P/P0 > 0.8) of both adsorption curves can be attributed to capillary condensation and multilayer adsorption of N2 in the mesopores or macropores. The hierarchical pore structures are also confirmed by pore size distributions of CN and ACN (Fig. 2b), which depict multi-modal pore structures expressly in the scope of 0.2–10 nm. The specific surface area of ACN is calculated to be 705.9 m2 g−1, which is higher than that of CN (212.4 m2 g−1). The results indicate both samples have similar porous structure, but the ACN sample possesses more abundant micropores and higher specific areas compared to CN sample.
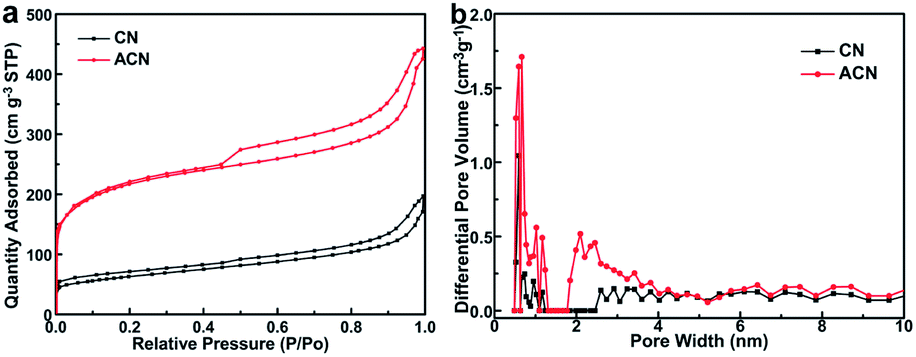 | ||
| Fig. 2 N2 adsorption–desorption isotherms (a) and pore size distributions (b) of CN and ACN. | ||
XPS analysis was conducted to determine the surface functionalities of CN and ACN as shown in Fig. 3. Fig. 3a shows the overall XPS survey spectra of the CN and ACN samples, which indicate that the obtained samples have rich nitrogen- and oxygen-containing functional groups without observing impurities. Nitrogen and oxygen contents of the ACN sample are as high as 19.8 wt% and 11.1 wt%, respectively, which are much higher than that of the CN sample (15.8 wt% and 4.0 wt%). High resolution spectral analysis of the N 1s and O 1s are conducted to determine their surface chemical binding states as shown in Fig. 3b–e. The N1s XPS spectrum of the CN sample (Fig. 3b) show six peaks at 398.1 (N1), 399.3(N2), 400.6(N3), 402.1(N4), 403.8(N5), and 406.1(N6) eV, which are assigned to pyridinic N, amine/amide, pyrrolic/pyridone N, quaternary N (N–Q), N-oxide/nitro, and chemisorbed N, respectively.30,33,46–48 The N1s XPS spectrum of the ACN sample (Fig. 3c) consists of five peaks except for chemisorbed N. The nitrogen contents, N/C ratio, and fitted results of N1s XPS spectra are calculated and summarized in Table S1 (see ESI†). The N1s spectrum changes point to a highly destructive effect from KOH treatment on the carbon framework. The nitrogen content of ACN is increased because carbon is consumed for the chemical activation reaction with KOH. Besides, the obtained carbon nanotubes derived from polyprrole precursor possesses more pyridinic, pyrrolic and quaternary N atomes (“lattice nitrogen”) rather than the thermal unstable “chemical nitrogen” under the pyrolysis conditions may be another reasonable explanation. The ACN sample has higher relative contributions of the pyrrolic/pyridone N forms (12.3 at%) compared to that of CN sample (6.3 at%), which are expected to lead to high electrochemical capacitance due to the pseudocapacitance effect.19,47,48 Fig. 3d and e present the O1s XPS spectra of the CN sample and the ACN sample, respectively. The O1s XPS spectra of the CN and ACN sample can be approximately fitted into four main peaks at ca. 530.6(O1), 531.5(O2), 532.4(O3) and 533.7(O4) eV, which represents C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O quinone type groups, CO or –COOH groups, C–O groups, and –OH groups.30,33,46–51 The oxygen contents, O/C ratio, and fitted results of O1s XPS spectra are calculated and summarized in Table S2 (see ESI†). The O/C ratio is highly increased after the KOH activation and the relative percentage of four oxygen functional groups is changed randomly. The obtained ACN possesses more abundant acidic functional sites, such as –COOH and –OH groups.
O quinone type groups, CO or –COOH groups, C–O groups, and –OH groups.30,33,46–51 The oxygen contents, O/C ratio, and fitted results of O1s XPS spectra are calculated and summarized in Table S2 (see ESI†). The O/C ratio is highly increased after the KOH activation and the relative percentage of four oxygen functional groups is changed randomly. The obtained ACN possesses more abundant acidic functional sites, such as –COOH and –OH groups.
 | ||
| Fig. 3 XPS survey spectra (a) of CN and ACN; high-resolution XPS spectra of N1s (b and c) and O1s (d and e) peaks of CN (b and d) and ACN (c and e). | ||
To evaluate the capacitive behaviour of both the CN and ACN samples, CV measurements were carried out with a three-electrode system in 1 mol L−1 H2SO4. Fig. 4a exhibits the CV curves of both CN and ACN electrodes at a scan rate of 5 mV s−1. The CV curve of the CN electrode shows a rectangular-like shape with an obvious well-broaden redox peak at lower potential (−0.2–0.6 V). While the ACN electrode presents capacitive behaviour with roughly rectangular-like shapes and a few humps owing to the synergic effects of EDLC and pseudocapacitance. The ACN electrode show more distinctive deviations of CV curves than that of the CN electrode, which indicating the contributions of pseudocapacitance from the ACN electrode are larger than that of the CN electrode. This may be due to the increased functional group proportion of N/C and O/C after the KOH activation. When the scan rate is increased to 50 mV s−1, the CV curve obtained from ACN electrode shows a unique rectangular-like shape and no dramatic distortions are observed as shown in Fig. 4b. However, the CN electrode exhibits dramatically distorted CV curve at the same scan rate (Fig. 4b). In addition, we investigate the rate-dependent CV curves for both electrodes over a range of scan rates from 5 to 100 mV s−1 (Fig. 4c and d). It can be seen clearly that even at a high scan rate of 100 mV s−1, a quasi-rectangular shape for the CV curves of the ACN electrode can be kept. In comparison with the CN electrode, a large deviation from the rectangular shape is observed in the case of the CN electrode due to the limitation in charge transfer processes at a relative high scan rate. Furthermore, the ACN electrode presents relatively larger area of CV curves compared to that of CN electrode at various scan rates, which are attributed to the coexistence of relatively larger surface area and more abundant interfacial functional groups. These nitrogen- and oxygen-containing functional groups are all advantageous. On one hand, the inductive effects of the s-bonded structure from O and N heteroatoms will cause a redistribution of the electrons as well as the polarization of some bonds, which can induce the electric potential redox reactions of these polarized sites through the reversible gaining/losing of electrons and simultaneous adsorption–desorption of protons, respectively. On the other hand, the acidic functional sites such as –COOH, –OH and pyrrolic N, may play the key role in reversible Faradic redox reactions (involving the reversible attachment/detachment of hydroxy groups between pyridone and pyridine as well as the oxidation of nitrogen atoms in pyridine).47,52–54 We calculate the specific capacitance of both CN and ACN electrodes from CV curves. Fig. S2† shows the variation in the specific capacitance as a function of scan rates. The specific capacitances of ACN electrode are 411.8, 398.7, 387.6, 358.4 and 325.4 F g−1 at scan rates of 5, 10, 20, 50 and 100 mV s−1, respectively. As for the CN electrode, the corresponding specific capacitances are 272.0, 258.4, 229.5, 166.6 and 105.1 F g−1 at scan rates of 5, 10, 20, 50 and 100 mV s−1, respectively.
 | ||
| Fig. 4 CV curves of both CN and ACN electrodes at a scan rate of 5 mV s−1 (a) and 50 mV s−1 (b); CV curves of CN (c) and ACN (d) electrodes over a range of scan rates from 5 to 100 mV s−1. | ||
The capacitive performances for both the CN and ACN samples were further tested with galvanostatic charge–discharge experiments. Fig. 5a and b depict the typical charge–discharge curves of the CN and ACN electrodes at 0.5 and 10 A g−1, respectively. The discharge curves of both the CN and ACN samples are in distorted linear shapes at 0.5 A g−1, as shown in Fig. 5a, which imply the operation of pseudocapacitance for both electrodes due to their high heteroatom doping level. However, the deviations in galvanostatic charge–discharge curves of the CN electrode are weaker; indicating the relative contribution of pseudocapacitance is smaller. There is no obvious ohmic drop even at a high current density of 10 A g−1 for ACN electrode (Fig. 5b), indicating that the ACN electrode can possess the small equivalent series resistance and good capacitive performance during the rapid charging–discharging process. Fig. 5c and d depict the galvanostatic charge–discharge curves of the CN and ACN samples over a range of current densities from 2 to 50 A g−1, respectively. For the CN electrode (Fig. 5c), although the charge–discharge curves retain a quasi-triangular shape at low current density, they are out of their shape and asymmetric when evaluated at a high current density. All charge–discharge curves (Fig. 5d) of the ACN sample at various current densities are quasi-triangular and symmetrical, suggesting that the electrode possesses typical EDLC behaviour and part of pseudocapacitive behaviour. The specific capacitances of both electrode materials were calculated from galvanostatic discharge curves, and the comparison of the capacitance retention for both samples at a current density range form 0.5 to 20 A g−1 is presented in Fig. 5e. At a current density of 0.5 A g−1, specific capacitances of 246.3 and 384.9 F g−1 are obtained for the CN and ACN electrodes, respectively. These results are in accordance with the value from CV curves. It is found that specific capacitance decreases with the increase of current density, which is related to the increase of diffusion limitation. At a high current density of 20 A g−1, the specific capacitance of ACN electrode is still maintained at 227.5 F g−1, which is much higher than that of CN electrode (60 F g−1). The ACN electrode delivers 201 F g−1 even at a high current density of 50 A g−1. The results indicate the ACN electrode exhibits superior capacitive performance, especially high rate performance and reversibility, compared to the CN electrode and other related samples reported previously.12,15,18,19,51 The excellent capacitive performance of the ACN electrode also should be attributed to its abundant micropores and high specific areas, which provide fast electrolyte diffusion pathways, low ion-transport resistance as well as good electrolyte penetration. The cyclic stability of the capacitance performances of both CN and ACN electrodes were evaluated by examination of 500 cycles at a consecutive high charging-discharging current of 10 A g−1 (Fig. 5f). The specific capacitances of the ACN electrode firstly drop by ca. 2.4% from 247.2 to 241.2 F g−1 during the first 10 cycles, and then remain almost constant for the rest cycles, indicating excellent long-term cycle ability and a high degree of reversibility in consecutive charge–discharge cycles. To understand the facilitated ion and electron transport behaviour of both electrodes, EIS tests were carried out and the results are shown in Fig. S3 (see ESI†). Both the CN and ACN electrodes exhibit a semicircle in the high frequency region and a sloping line at low frequencies. Compared with the CN electrode, the ACN electrode shows the straightest line with an almost 90°, which is characteristic of better capacitive behaviour. In addition, the charge transfer resistance (Rct) was calculated to be 9.2 and 1.2 Ω for the CN and ACN electrodes, respectively. The results demonstrate the ACN electrode indeed shows facilitated ion and electron transport behaviour owing its abundant micropores, high specific areas, and abundant interfacial functional groups.
 | ||
| Fig. 5 Galvanostatic charge–discharge curves of CN and ACN electrodes at a current density of 0.5 A g−1 (a) and 20 A g−1 (b); (c) galvanostatic charge–discharge curves of CN (c) and ACN (d) electrode over a range of current densities from 2 to 50 A g−1; (e) the specific capacitances from galvanostatic charge–discharge curves versus discharge current density; (f) cycling performances of CN and ACN electrodes at a current density of 10 A g−1. | ||
Conclusions
Nitrogen- and oxygen-containing ACN with an enlarged specific area of 705.9 m2 g−1, a high nitrogen and oxygen contents of 19.8 wt% and 11.1 wt%, have been successfully synthesized by a high temperature carbonization of PPy nanotubes followed by chemical activation. The abundant micropores and high specific areas of ACN are advantageous for EDLCs. The abundant functional groups contribute to pesudocapacitance. The obtained ACN electrode exhibits enhanced capacitance performances, such as high specific capacitance (384.9 F g−1 at 0.5 A g−1), excellent rate capability (201 F g−1 at 50 A g−1) and more stable cyclic stability (only 2.4% of specific capacitance loss after 500 cycles). The finding on ACN with engineering of surface area and composition are expected to open up new opportunities in the design of high-performance carbon electrode materials for supercapacitors.Acknowledgements
This work was supported by the National Natural Science Foundation of China (51272113), the Natural Science Foundation of Shandong Province (2009ZRB01034), and Open Foundation of State Key Laboratory of Coal Conversion, China (J12-13-903).Notes and references
- J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef CAS PubMed.
- C. Largeot, C. Portet, J. Chmiola, P. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730 CrossRef CAS PubMed.
- M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS.
- J. R. Miller, R. A. Outlaw and B. C. Holloway, Science, 2010, 329, 1637 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- E. Frackowiak and F. Béguin, Carbon, 2001, 39, 937 CrossRef CAS.
- Y. Korenblit, M. Rose, E. Kockrick, L. Borchardt, A. Kvit, S. Kaskel and G. Yushin, ACS Nano, 2010, 4, 1337 CrossRef CAS PubMed.
- L. Wei, M. Sevilla, A. B. Fuertes, R. Mokaya and G. Yushin, Adv. Funct. Mater., 2012, 22, 827 CrossRef CAS.
- Y. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537 CrossRef CAS PubMed.
- M. S. Balathanigaimani, W. G. Shim, M. J. Lee, C. Kim, J. W. Lee and H. Moon, Electrochem. Commun., 2008, 10, 868 CrossRef CAS PubMed.
- D. W. Wang, F. Li, M. Liu, G. Q. Lu and H. M. Cheng, Angew. Chem., Int. Ed., 2008, 47, 373 CrossRef CAS PubMed.
- F. B. Su, C. K. Poh, J. S. Chen, G. W. Xu, D. Wang, Q. Li, J. Y. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717 CAS.
- A. Izadi-Najafabadi, T. Yamada, D. N. Futaba, M. Yudasaka, H. Takagi, H. Hatori, S. Iijima and K. Hata, ACS Nano, 2011, 5, 811 CrossRef CAS PubMed.
- A. Izadi-Najafabadi, S. Yasuda, K. Kobashi, T. Yamada, D. N. Futaba, H. Hatori, M. Yumura, S. Iijima and K. Hata, Adv. Mater., 2010, 22, E235 CrossRef CAS PubMed.
- H. Wang, Y. Wang, Z. Hu and X. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6827 CAS.
- Y. Zhao, J. Liu, Y. Hu, H. Cheng, C. Hu, C. Jiang, L. Jiang, A. Cao and L. Qu, Adv. Mater., 2013, 25, 591 CrossRef CAS PubMed.
- X. W. Yang, J. W. Zhu, L. Qiu and D. Li, Adv. Mater., 2011, 23, 2833 CrossRef CAS PubMed.
- L. Sun, L. Wang, C. G. Tian, T. X. Tan, Y. Xie, K. Y. Shi, M. T. Li and H. G. Fu, RSC Adv., 2012, 2, 4498 RSC.
- Y. Tan, C. Xu, G. Chen, Z. Liu, M. Ma, Q. Xie, N. Zheng and S. Yao, ACS Appl. Mater. Interfaces, 2013, 5, 2241 CAS.
- Z. S. Wu, Y. Sun, Y. Z. Tan, S. Yang, X. Feng and K. Müllen, J. Am. Chem. Soc., 2012, 134, 19532 CrossRef CAS PubMed.
- T. Bordjiba, M. Mohamedi and L. H. Dao, Adv. Mater., 2008, 20, 815 CrossRef CAS.
- Y. Li, Z. Li and P. K. Shen, Adv. Mater., 2013, 25, 2474 CrossRef CAS PubMed.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930 CAS.
- A. E. Fischer, K. A. Pettigrew, D. R. Rolison, R. M. Stroud and J. W. Long, Nano Lett., 2007, 7, 281 CrossRef CAS PubMed.
- D. Liu, X. Wang, X. Wang, W. Tian, J. Liu, C. Zhi, D. He, Y. Bando and D. Golberg, J. Mater. Chem. A, 2013, 1, 1952 CAS.
- X. Wang, J. S. Lee, Q. Zhu, J. Liu, Y. Wang and S. Dai, Chem. Mater., 2010, 22, 2178 CrossRef CAS.
- M. M. Titirici, A. Thomas and M. Antonietti, J. Mater. Chem., 2007, 17, 3412 RSC.
- M. Sevilla, L. Yu, T. P. Fellinger, A. B. Fuertes and M. M. Titirici, RSC Adv., 2013, 3, 9904 RSC.
- M. Sevilla, R. Mokaya and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 2930 CAS.
- B. Xu, D. Zheng, M. Jia, G. Cao and Y. Yang, Electrochim. Acta, 2013, 98, 176 CrossRef CAS PubMed.
- W. Gu, M. Sevilla, A. Magasinski, A. B. Fuertes and G. Yushin, Energy Environ. Sci., 2013, 6, 2465 CAS.
- D. Zhang, Y. Hao, L. Zheng, Y. Ma, H. Feng and H. Luo, J. Mater. Chem. A, 2013, 1, 7584 CAS.
- D. H. Jurcakova, M. Seredych, G. Q. Lu and T. J. Bandosz, Adv. Funct. Mater., 2009, 19, 438 CrossRef.
- E. Raymundo-Piñero, F. Leroux and F. Béguin, Adv. Mater., 2006, 18, 1877 CrossRef.
- H. Zhu, X. Wang, X. Liu and X. Yang, Adv. Mater., 2012, 24, 652 Search PubMed.
- P. Chen, T. Y. Xiao, Y. H. Qian, S. S. Li and S. H. Yu, Adv. Mater., 2013, 25, 3192 CrossRef CAS PubMed.
- L. Qie, W. M. Chen, Z. H. Wang, Q. G. Shao, X. Li, L. X. Yuan, X. L. Hu, W. X. Zhang and Y. H. Huang, Adv. Mater., 2012, 24, 2047 CrossRef PubMed.
- L. F. Chen, X. D. Zhang, H. W. Liang, M. Kong, Q. F. Guan, P. Chen, Z. Y. Wu and S. H. Yu, ACS Nano, 2012, 6, 7092 CrossRef CAS PubMed.
- D. W. Wang, F. Li, J. Zhao, W. Ren, Z. G. Chen, J. Tan, Z. S. Wu, I. Gentle, G. Q. Lu and H. M. Cheng, ACS Nano, 2009, 3, 1745 CrossRef CAS PubMed.
- H. Marsh and F. R. Reinoso, Activated Carbon, Elsevier, Oxford, 2006 Search PubMed.
- D. Hulicova, M. Kodama and H. Hatori, Chem. Mater., 2006, 18, 2318 CrossRef CAS.
- Y. Chen, X. Zhang, D. Zhang, P. Yu and Y. Ma, Carbon, 2011, 49, 573 CrossRef CAS PubMed.
- C. Guo, N. Li, L. Ji, Y. Li, X. Yang, Y. Lu and Y. Tu, J. Power Sources, 2014, 247, 660 CrossRef CAS PubMed.
- G. Xu, B. Ding, P. Nie, L. Shen, J. Wang and X. Zhang, Chem. – Eur. J., 2013, 19, 12306 CrossRef CAS PubMed.
- S. Shang, X. Yang and X. Tao, Polymer, 2009, 50, 2815 CrossRef CAS PubMed.
- R. Arrigo, M. Hävecker, S. Wrabetz, R. Blume, M. Lerch, J. McGregor, E. P. J. Parrott, J. A. Zeitler, L. F. Gladden, A. Knop-Gericke, R. Schlögl and D. S. Su, J. Am. Chem. Soc., 2010, 132, 9616 CrossRef CAS PubMed.
- Q. Kong, C. Chen, Q. Zhang, X. Zhang, M. Wang and R. Cai, J. Phys. Chem. C, 2013, 117, 15496 CAS.
- G. Tian, M. Zhao, Q. Zhang, J. Huang and F. Wei, Carbon, 2012, 50, 5323 CrossRef CAS PubMed.
- D. W. Wang, F. Li, L. C. Yin, X. Lu, Z. G. Chen, I. R. Gentle, G. Q. Lu and H. M. Cheng, Chem. – Eur. J., 2012, 18, 5345 CrossRef CAS PubMed.
- X. Yang, D. Wu, X. Chen and R. Fu, J. Phys. Chem. C, 2010, 114, 8581 CAS.
- S. Biniak, G. Szymanski, J. Siedlewski and A. Swiatkowski, Carbon, 1997, 35, 1799 CrossRef CAS.
- C. Chen, Q. Zhang, X. Zhao, B. Zhang, Q. Kong, M. Yang, Q. Yang, M. Wang, Y. Yang, R. Schlögl and D. S. Su, J. Mater. Chem., 2012, 22, 14076 RSC.
- D. Wang, F. Li, L. Yin, X. Lu, Z. Chen, I. R. Gentle, G. Q. Lu and H. Cheng, Chem. – Eur. J., 2012, 18, 5345 CrossRef CAS PubMed.
- C. Chen, Q. Zhang, M. Yang, C. Huang, Y. Yang and M. Wang, Carbon, 2012, 50, 3571 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM and TEM images, Nyquist plots, and tables. See DOI: 10.1039/c3ra45076g |
| This journal is © The Royal Society of Chemistry 2014 |