 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rational design of the electrode morphology for oxygen evolution – enhancing the performance for catalytic water oxidation
Aleksandar R. Zeradjanin*a,
Angel A. Topalova,
Quentin Van Overmeereb,
Serhiy Cherevkoa,
XingXing Chenc,
Edgar Ventosac,
Wolfgang Schuhmannc and
Karl J. J. Mayrhofer*a
aDepartment of Interface Chemistry and Surface Engineering, Max-Planck-Institut für Eisenforschung GmbH, Max-Planck-Strasse 1, 40237 Düsseldorf, Germany. E-mail: zeradjanin@mpie.de; mayrhofer@mpie.de
bInstitute of Mechanics, Materials and Civil Engineering, Université catholique de Louvain, Place sainte-Barbe 2, B-1348 Louvain-la-Neuve, Belgium
cAnalytische Chemie – Elektroanalytik & Sensorik, Ruhr-Universität Bochum, Universitätsstr. 150, 44780 Bochum, Germany
First published on 27th January 2014
Abstract
The fundamental understanding of the electrode/electrolyte interface is of pivotal importance for the efficient electrochemical conversion and storage of electrical energy. However, the reasons for the low rate of electrocatalytic oxygen evolution and issues of long-term material stability, which are central constraints for attaining desirable efficiency for sustainable technologies like water electrolysis or electrochemical CO2 reduction, are still not completely resolved. While a lot of attention has been directed towards the search for new materials with unique (electro)catalytic properties, experimental results accumulated during the last four decades and prediction from models suggest that RuO2 possesses superior activity for oxygen evolution under acidic conditions. Considering that RuO2 is a material of choice, we show that tailoring the surface morphology on the meso- and macroscale has great potential for the improvement of the efficiency of this gas evolving reaction. Advanced analytical tools have been utilized for the combined investigation of both activity and stability. Namely, the potential dependent frequencies of gas-bubble evolution, an indicator for the activity of the electrode, were acquired by scanning electrochemical microscopy (SECM), while the dissolution of RuO2 was monitored using a micro electrochemical scanning flow cell combined with an inductively coupled plasma mass spectrometer (SFC-ICP-MS). The obtained fundamental insights will aid improving the design and thus performance of electrode materials for water oxidation.
1. Introduction
Recently, the production of chemicals for energy storage (hydrogen, methanol, methane…) has received a lot of attention as an integral part of a renewable energy concept.1–6 The (electro)chemical energy conversion conceptually relies on postulates of (electro)catalysis, which are typically based on the impact of the nature of the electrode material on the rate of the reaction.7 Considering the limited amount of available mineral resources,3 additional aspects of crustal abundance, price and especially long-term stability of the materials are of at least equal importance within the concept of catalyst performance. The most substantial barrier towards targeted efficiencies for cathodic production of hydrogen (and methanol or methane) is still the high energy demand for electrolysis, particularly the high overpotential during the counter oxygen evolution reaction (OER).8 Moreover, the highly corrosive conditions during anodic water oxidation are a crucial factor for the material stability and thus the long-term performance, which will certainly affect the implementation of new technologies.9The high overpotential for the OER originates in the necessity to exchange four electrons during the (electro)catalytic process and the formation of at least three adsorbed intermediates.10 The preferences of different materials for the adsorption/desorption of these oxygen containing intermediates are believed to be the determining factors for the reaction rate. Several catalytic descriptors (for example, binding energies of key intermediates, O*, OH*, and OOH*) have been used to theoretically predict and explain the extensively studied activity trends, which are usually represented in the form of so-called “volcano” plots.11–14 The general perception is that RuO2 is positioned at or close to the top of the “volcano”, since it does not bind the reaction intermediates too strongly nor too weakly, so that according to the Sabatier principle the kinetics for generation of molecular oxygen during the water oxidation is at a maximum.12
Besides the OER activity, a key challenge to be addressed in the evaluation of electrocatalyst performance is the stability of the electrode material under relevant operational conditions. The degradation of materials used for electrocatalytic OER is strongly influenced by the pH, applied potentials/currents, impurities etc.15 At the highly positive potentials during the OER, dissolution of electroactive material is a detrimental process that has to be considered for any catalyst material.
A crucial factor for the performance of catalyst materials is the evolution of gases during the reaction. Namely, at respectable reaction rates the interfacial concentration of oxygen molecules becomes so high that the water at the electrode/electrolyte interface is not sufficient to create solvation spheres around each produced oxygen molecule. This phenomenon known as supersaturation is causing the formation of gas-bubbles at the interface.16 The effective detachment of these gas-bubbles is essential for the mass transport in a reactor.17,18 In contrast, the adhesion of gas-bubbles at the electrode surface blocks fractions of the active surface area and thereby induces additional overpotential. Thus the overall efficiency of the detachment of gas-bubbles during gas evolving reactions is at least of equal significance as the electrocatalysis of the reactions.7 The rational strategy in promotion of gas-bubble detachment relies on tailoring of the electrode surface morphology, which has proven to be a very efficient approach in accelerating the anodic gas evolution in the case of chlorine evolution reaction (CER).19,20 As for a series of transition metal-oxides the CER and the water oxidation exhibited the same activity trend, i.e. for the same current density the potential of the CER and water oxidation are linearly scaled with a slope equal to one,21 and respecting the fact that gas-bubble detachment is a process predominantly driven by the surface physics, it is justified to assume that surface morphology will influence both reactions in a similar manner.
In this study we investigate the impact of the surface morphology on the OER activity and the catalyst stability of RuO2 samples. The structure–activity relationship for water oxidation is established by the employment of a recently developed analytical approach based on the scanning electrochemical microscopy (SECM). Namely, considering that oxygen evolution during water oxidation is an oscillatory process, the SECM microelectrode tip was used as a sensor for measuring the frequency of gas-bubble detachment.22,23 The dissolution behavior of RuO2 during the OER is monitored with a scanning flow cell and online inductively coupled plasma mass spectrometer (SFC-ICPMS).24,25
2. Experimental
Conductive atomic force microscopy (c-AFM) was used to obtain information about the morphological pattern and about the spatial distribution of conductivity. c-AFM measurements were performed in contact mode in air using Pt/Ir coated silicon cantilevers from AppNano, USA (ANSCM-PC ∼ 12 kHz, ∼0.2 N m−1) with a NanoWizard III (JPK, Berlin, Germany). X-ray photoelectron spectroscopy (XPS) measurements were performed using a spectrometer PHI Quantum 2000 (Physical Electronics, USA) with a source analyzer angle of 45°. The base pressure in the spectrometer was 2 × 10−8 mbar. X-rays were obtained using a monochromatic AlKα source (energy of 1486.6 eV) at pass energy of 23.5 eV, and a energy resolution of 0.2 eV. The position of the carbon peak C 1s was at 284.1 eV. To avoid contamination, the samples were sputtered with an argon plasma with an energy of 1000 eV. An energy neutralizer was used to eliminate charging of the surface. The obtained XPS spectra were compared with spectra available in literature.26A special configuration of the scanning electrochemical microscopy (SECM) was adopted in order to estimate the frequency of gas-bubble detachment. An aqueous solution of H2SO4 (0.1 M, 99.5%, J. T. Baker, Deventer, Netherlands) was used as electrolyte solution. All electrolytes were prepared by using de-ionized water (SG Ultra Clear UV, Barsbüttel, Germany). The SECM tip was a Pt microelectrode with a diameter, dt, of 25 μm. The reference electrode was an Ag/AgCl/3M KCl electrode [0.207 V vs. a normal hydrogen electrode (NHE)], and the counter electrode was a Pt mesh. Before each measurement, the microelectrode was polished with 0.3 μm alumina paste, cleaned in an ultrasonic bath, and rinsed with acetone and water. The Pt wires, the Pt mesh, and Ag wires were provided by Goodfellow (Bad Nauheim, Germany). SECM experiments were performed by using a four-electrode system consisting of two WEs (sample and tip), a RE, and a CE controlled by a bipotentiostat/galvanostat (Jaissle PG 100, Waiblingen, Germany). The position of the tip was adjusted in the x, y, and z directions by a stepmotor-based SECM system (Sensolytics; Bochum, Germany). Prior to measurements, the microelectrodes were characterized by cyclic voltammetry to control their surface status. The feedback mode of the SECM was used to record the approach curves and pre-position the SECM tip at a distance h, which was approximately 1 μm from the sample surface. The potential applied to the tip was 400 mV, while the sample potential was adjusted in the range from OCP till 1.8 V vs. Ag/AgCl/3M KCl with increments of 100 mV. The current at the tip was monitored with a frequency of 100 Hz over 60 s. All measurements were performed at 25 °C.
The micro-electrochemical scanning flow cell (SFC) was described in detail before.27 The microcell was mounted on a force sensor (MEMesssysteme, Germany) that adjusts the force to 400 ± 5 mN in contact mode. The contact area of the electrolyte on the substrate was thus reproducibly 1.1 mm2. The sample positioning is achieved by a XYZ-translation stage (Physik Instrumente, Germany). An in-house developed LabVIEW program controls all hardware components simultaneously.28 Electrochemical measurements were performed with a potentiostat (Gamry Reference 600). The electrolyte pumped through the microcell and over the working electrode is subsequently introduced into the inductively coupled plasma mass spectrometer (ICP-MS, NexION 300X, Perkin Elmer), equipped with a cyclonic spray chamber and a Meinhard concentric nebulizer. The RF power for the plasma generation was held at 1300 W with argon flow of 15 L min−1. The dissolution rate of ruthenium was monitored over the two isotopes 102Ru and 104Ru with respect to 5 μg L−1 internal standard 89Y, mixed into the electrolyte directly after the SFC with a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The acquisition parameters of the ICP-MS were set to 100 ms dwell time and 5 sweeps per reading. The detected intensities were quantitatively evaluated using a calibration of the ICP-MS for the Ru isotopes, which was performed prior to each measurement.
:1. The acquisition parameters of the ICP-MS were set to 100 ms dwell time and 5 sweeps per reading. The detected intensities were quantitatively evaluated using a calibration of the ICP-MS for the Ru isotopes, which was performed prior to each measurement.
3. Design and synthesis of catalyst
The rational design of an electrode surface requires a certain theoretical basis that can help us to establish a more exact link between synthesis procedures and desired properties. Although it is usually assumed that catalyst coatings have relatively uniform distribution of active sites, surface inhomogeneities can be quite severe.19 Reducing surface inhomogeneities is therefore recognized as one of the beneficial perspectives for the design of electrodes for efficient gas evolution.29,30 Moreover, based on our previous works,19,22 the morphological pattern of the electrode surface is crucial for the nucleation, limited growth and fast detachment of gas-bubbles. In particular, evenly distributed “cracks” having a size that corresponded to the critical diameter of the gas-bubble nucleation can enhance the activity significantly.19,22The formation of “cracks” in a catalytic layer during synthesis is typically achieved by inducing a large enough tensile stress in the layer.31 During sol–gel synthesis a wet layer of salt precursors of the catalyst is deposited on a solid substrate. After drying, the catalyst is exposed to thermal annealing to transform it into the active state. The volume change of the gel-body during the drying and thermal treatment steps is restricted by the substrate, which results in an internal stress that depends on the density change of the coating during drying and the mismatch in the coefficients of thermal expansion between thin coating and thick substrate. If the internal stress is large enough, surface cracks or channel cracks may nucleate and release energy.32 The internal stress σ in a thin coating can be estimated by:
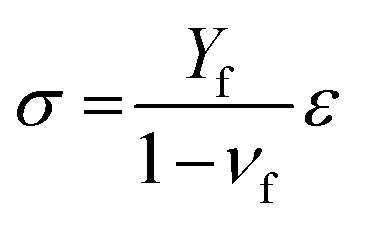
| ε = εdrying + (αf − αs)(T0 − T) |
where Γ is the fracture resistance of the coating and Z is equal to 3.951 for surface cracks and 1.976 for channel cracks. For brittle coatings on compliant substrates, the separation between cracks is found to decrease proportionally to 1/σ2.32,33
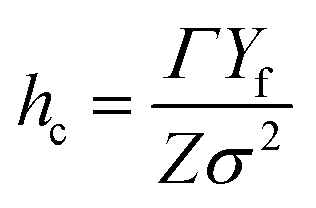
From the relations shown above, different morphologies can be obtained by varying the synthesis parameters such as the synthesis temperature, coating composition (thus modifying αf) or substrate composition (this modifying αs). In this initial study on the OER on RuO2 we focused on two clearly distinctive cases, a cracked electrode with a targeted “crack” size that corresponds to the critical diameter of gas-bubble nucleation, and a crack-free electrode morphology, in order to obtain first insight into the performance differences. The synthesis of the samples was done according to a previously reported procedure based on a sol–gel route.31 Briefly, Ti substrates (diameter 15 mm, Goodfellow, USA) were drop-coated with a mixture of adequate precursor solutions. The wet coated surface was dried in air and thermally treated until a mixed oxide of RuOx/SnOx was formed. The procedure was repeated until a Ru loading of 5.78 g m−2 (metal basis) was achieved. To prepare a “crack-free” coating with the same loading, a diluted sol solution (diluted in 1:31 ratio in isopropanol) was used and the number of the coating–drying–sintering cycles was increased accordingly. The different morphologies result from the deposited layer thickness during each step being larger and smaller than the critical thickness for the cracked and crack-free morphologies, respectively. The synthesized electrodes were of the dimensionally stable anodes type (DSA),7 with RuOx as the active component and SnOx as the matrix with a similar crystal lattice to the active component.34 The role of the SnOx matrix is to improve the stability of the catalytic component, while SnOx itself is electrocatalytically inactive for the OER.35 Dependent on the details of the thermal treatment, the obtained phase was predominantly rutile.31
4. Results and discussion
4.1. Surface characterization (morphology, composition and number of active sites)
In Fig. 1 the two targeted morphological patterns obtained during the synthesis can be clearly distinguished. The “cracked” surface, made out of a relatively small number of coating cycles, contains “terraces” and “cracks”, while the “crack-free” surface, made out of larger number of coating cycles with a diluted coating solution, exhibits agglomeration of active material and microcluster formation, similar to the coatings previously obtained via electrochemical deposition.20 The “crack-free” surface is a typical example of an on-top deposited catalytically active material, manifested in particles or clusters distributed randomly at the electrode surface. In contrast, the “cracked” surface has unique properties due to terraces of a width of ca. 10 μm in diameter. The exposed edges of the “terraces” are regions where locally very high activities can be obtained, concomitantly causing high local current density. At the same time, “cracks” can behave like channels that enable certain specific internal hydraulic regime. The conductivity distribution in Fig. 1 is well in relation with the morphological pattern. Note that the obtained conductivity values are presented on a relative scale, nevertheless they are sufficiently descriptive to illustrate local surface heterogeneities. While for the “crack-free” sample the most conductive regions are microclusters, in the case of “cracked” electrodes the most conductive regions are mainly situated close to “cracks” or more precisely at the edges of the “terraces”.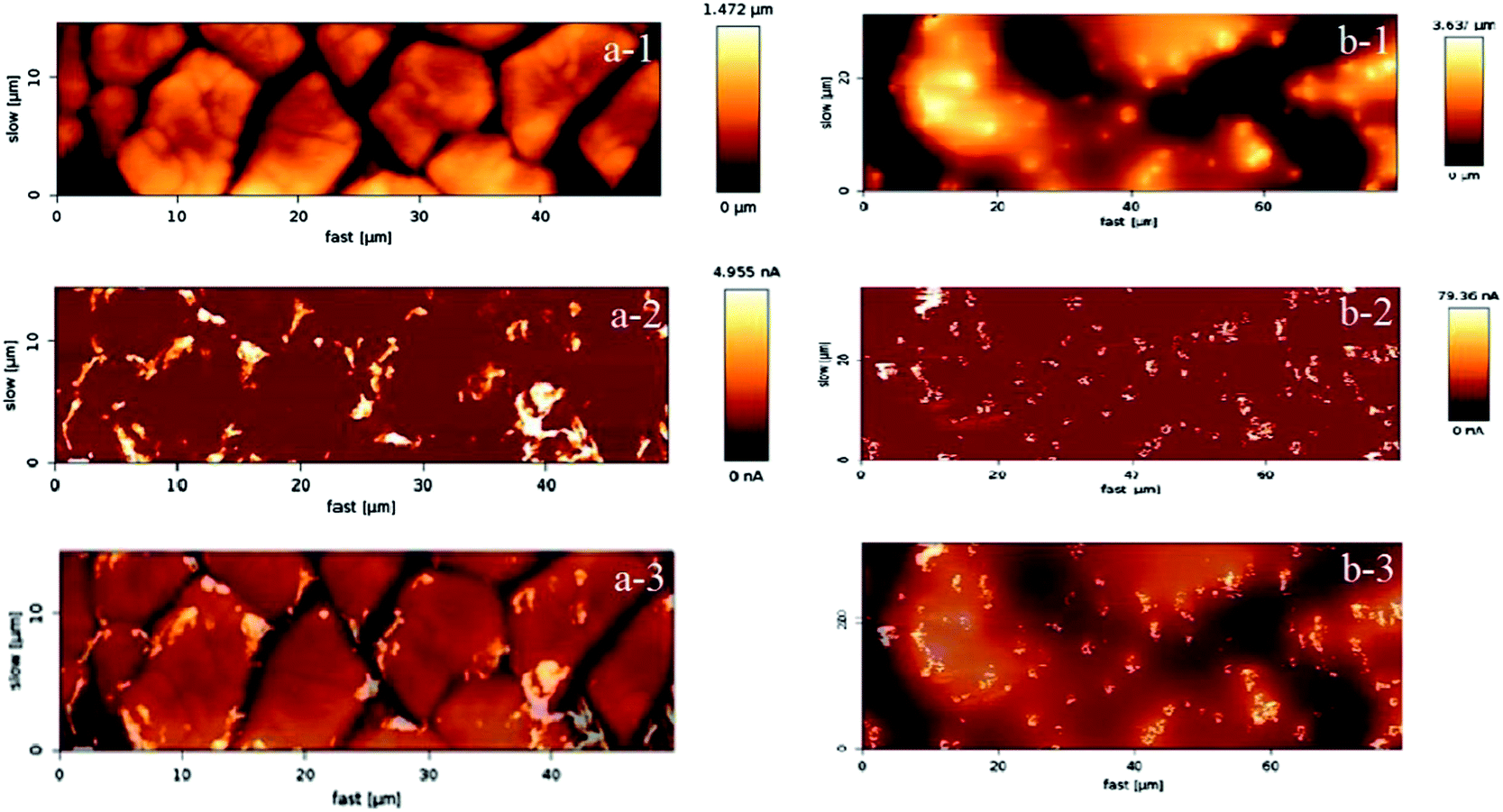 | ||
| Fig. 1 AFM micrographs of (a) the “cracked” electrode in tapping mode (a-1), conductive mode (a-2) and overlapped tapping and conductive mode (a-3); (b) the “crack-free” electrode in tapping mode (b-1), conductive (b-2) mode and overlapped tapping and conductive mode (b-3). | ||
To confirm that predominantly the geometric properties (morphology and active surface area) influence the efficiency of the gas-evolution, the samples should have relatively similar chemical behavior. The similar features for Ru, Sn and O in the wide-scan XPS spectra (Fig. 2a) of both samples supports this, although a complete quantitative overlap is missing.36
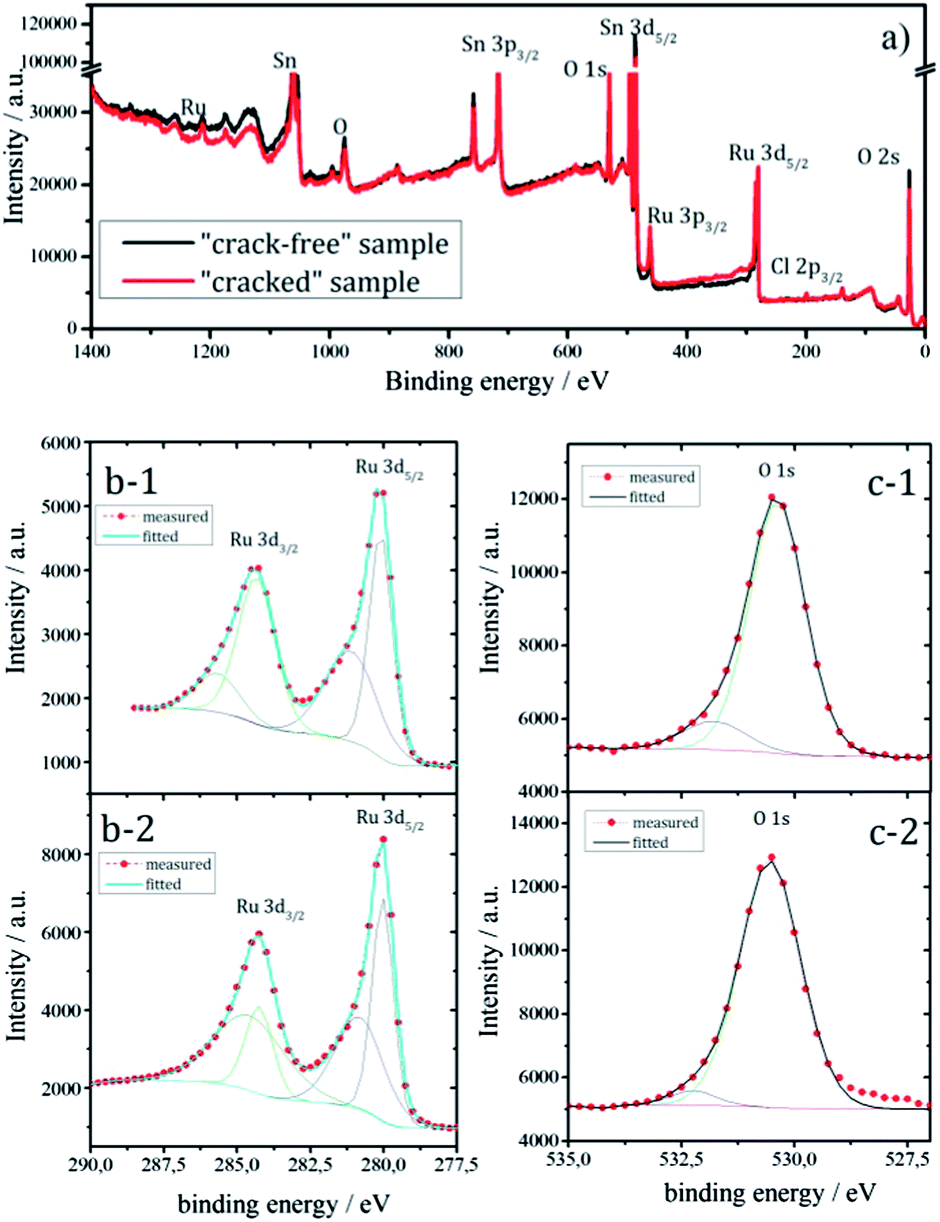 | ||
| Fig. 2 (a) Wide-scan XPS spectra of the “crack-free” (black) and “cracked” (red) sample after ion sputtering, (b) XPS spectra of the Ru 3d core level for “crack-free” (1) and “cracked” (2) sample, (c) XPS spectra of the O 1s level for “crack-free” (1) and “cracked” (2) sample. | ||
Since Ru centers are a key for the catalytic performance, it is important to gain an insight about the amount of Ru (usually various nonstoichiometric oxides RuOx where x < 2) at near surface regions. The relative intensities in Fig. 2b suggest that the “cracked” sample contains more Ru than the “crack-free” sample at near surface regions. At the same time it seems from the O 1s level (Fig. 2c) that the oxygen content in both samples is similar. Note that the XPS spectra (Fig. 2b and c) are recorded after sputtering, which minimizes the interference of the carbon C 1s peak (binding energy of 284.3 eV) on Ru 3d, but also Ru-oxides can be partially reduced. Similar information on the Ru content with respect to Sn and O was obtained from EDX mapping (not shown): the “cracked” sample (11.5 at%) has a higher content of Ru than the “crack-free” sample (8.5 at%). This relatively small difference in the Ru content is not expected to have a significant influence on the intrinsic catalytic properties of the electrode as shown previously.19,37 In the mentioned works the established activity trends could not be related with the loading, Ru content or overall electrochemical active surface area.
While XPS provides an indication of the average surface composition for several nanometers of the outer surface of the catalyst layer, only the outermost atomic layer that is directly in the contact with the electrolyte is relevant for electrochemistry. Thus the surface composition has to be obtained ideally in situ, utilizing for instance cyclic voltammetry (CV).38 The Brønsted type acid/base behavior of RuOx and its capability to reversibly exchange protons can be used to estimate the electrochemically active area:39–41
| RuOx(OH)y + zH+ + e− ⇆ RuOx−z(OH)y+z |
Examples of CV's of the analyzed samples are shown in the inset of Fig. 3. The integral of the area of the CV's divided by the potential scan rate gives the voltammetric surface charge, which corresponds to the electrochemically active surface area or number of active sites on the observed geometric region.
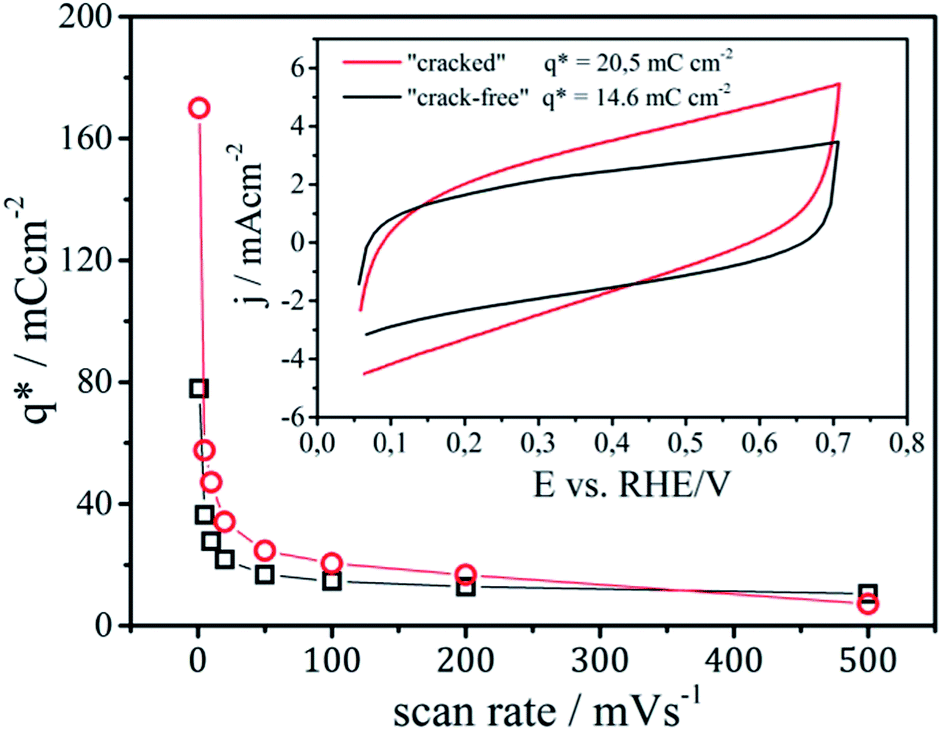 | ||
| Fig. 3 Dependence of the voltammetric surface charge on the potential scan rate. Inset: cyclic voltammograms of RuOx-based samples in 0.1 M H2SO4 recorded at a scan rate of 100 mV s−1; values of voltammetric surface charge corresponding to the electrochemically active surface area are indicated. | ||
The exchange of protons between the electrolyte and Ru-centers from the catalyst layer is a time dependent diffusion limited process. Therefore, an increase of the voltammetric surface charge is observed with the decrease of the scan rate, which is more pronounced in the case of the “cracked” sample. Two regions can be distinguished in Fig. 3, one corresponding to the “outer” surface area which can be probed at higher scan rates and one to the “inner” surface area of the porous coatings which can be quantified only at low scan rates (ideally on a scan rate equal to zero).31 The formal point of distinction between an “outer” and an “inner” surface area is its accessibility.42,43 The easily accessible “outer” surface area is considered to participate in the reaction,43 while the “inner” surface area that predominantly originates from nanopores is less or not accessible for the reaction.44 The “outer” surface area for the “cracked” and the “crack-free” sample is almost identical with around 10 mC cm−2. However, the “cracked” sample contains a significantly higher “inner” surface area, due to the fact that “cracked” electrodes are more porous.42 Practically, at a scan rate of 1 mV s−1 (in a first approximation considered to be steady state) the surface area of the “cracked” sample is more than two times larger than the area of the “crack-free” sample.
Note in this context, that while it was usually considered that only the “outer” surface area participates actively in the electrode reaction,20,43 it was suggested by Evdokimov that at very high current densities (above 1 A cm−2) the porous parts of the catalytic layer start to participate in the reaction due to accelerated mass transport inside the pores.45,46 The imposed operation regime (in this case current density) can thus influence the effectiveness of the observed catalytic layer. At the same time it was assumed that an activation of the “inner” surface of the electrode at a defined regime of current density can be achieved by gradually changing the morphological pattern. Although gradual changing of the morphology (thickness, “crack” size etc.19,22,31) has an influence on the catalyst performance, only one type of cracked coating has shown a distinctively different behavior. This was due to the fact that the thickness and crack size of this particular sample was similar to the critical diameter of gas-bubble nucleation.19,22 Consequently, instead of gradually changing the surface morphology,19,22,31 in this work a more straightforward OER analysis of two extreme patterns was performed.
4.2. Specific activity for OER
In order to resolve the impact of the real and effective surface area for this gas-evolving reaction, and thereby estimate the accessibility of the active sites, the measured currents have to be normalized.47–49 The normalization of current with the voltammetric surface charge representing the surface area yields a true turnover frequency that reflects the intrinsic activity. As the “outer” surface area is approximately the same for both samples (around 10 mC cm−2), the ratio between the normalized activities does not change. When the current density is normalized for the “inner” surface area, however, the OER activity of both samples becomes comparable (see Table 1).| Sample | jgeo/mA cm−2 | q*out/mC cm−2 | q*in/mC cm−2 | jgeo/q*out/Hz | jgeo/q*in/Hz |
|---|---|---|---|---|---|
| CS | 15.5 | 10.0 | 170.1 | 1.55 | 0.091 |
| CFS | 8.5 | 10.0 | 77.8 | 0.85 | 0.11 |
In a first approximation these results suggest that the higher current intensity of the “cracked” sample can be due to the superior “inner” surface area and a complete accessibility of these sites for the reaction. This would confirm previous studies, where the behavior of ”cracked” morphological patterns was ascribed to the higher density of active sites in comparison to “crack-free” electrodes.42 In contrast, however, other studies on RuO2 based anodes have shown that a high density of active sites is not a guarantee for efficient performance during gas-evolving reactions.19,31 Certain “crack-free” electrodes can exhibit comparable or even higher surface area and enhanced nanoporosity to “cracked” electrodes at similar loadings, but still be less efficient.31 Considering all of this it is necessary to extend the analysis and find an approach that provides a deeper insight if indeed only the inner surface of the reaction, or if more efficient gas detachment from the “outer” surface is also strongly influencing the activity (see Section 4.4).
4.3. Evaluation of electrode stability
The electrode stability during OER is not only equally important for the catalytic performance as the activity, but can also aid the understanding of the impact of the effective surface area. While several deactivation mechanisms for the oxide electrodes are discussed in literature15 (detachment of the coating, influence of impurities, passivation of the support etc.), the most crucial from the (electro)chemical point of view is dissolution of the catalytically active component. The dissolution of elements is an integral part of the surface reconstructing during the OER and in that sense insights can help us to find means how to passivate and/or protect the electrode surface from degradation. Due to the similar chemistry, it is expected that the dissolution rates for the two samples will depend on the overpotential for the OER, which is strongly affected by the effective surface area and consequently by the surface morphology.The impact of the morphological pattern on the amount of dissolved ruthenium during an identical electrochemical test can be seen from the ICP-MS dissolution profiles depicted as a function of the applied potential and time in Fig. 4.
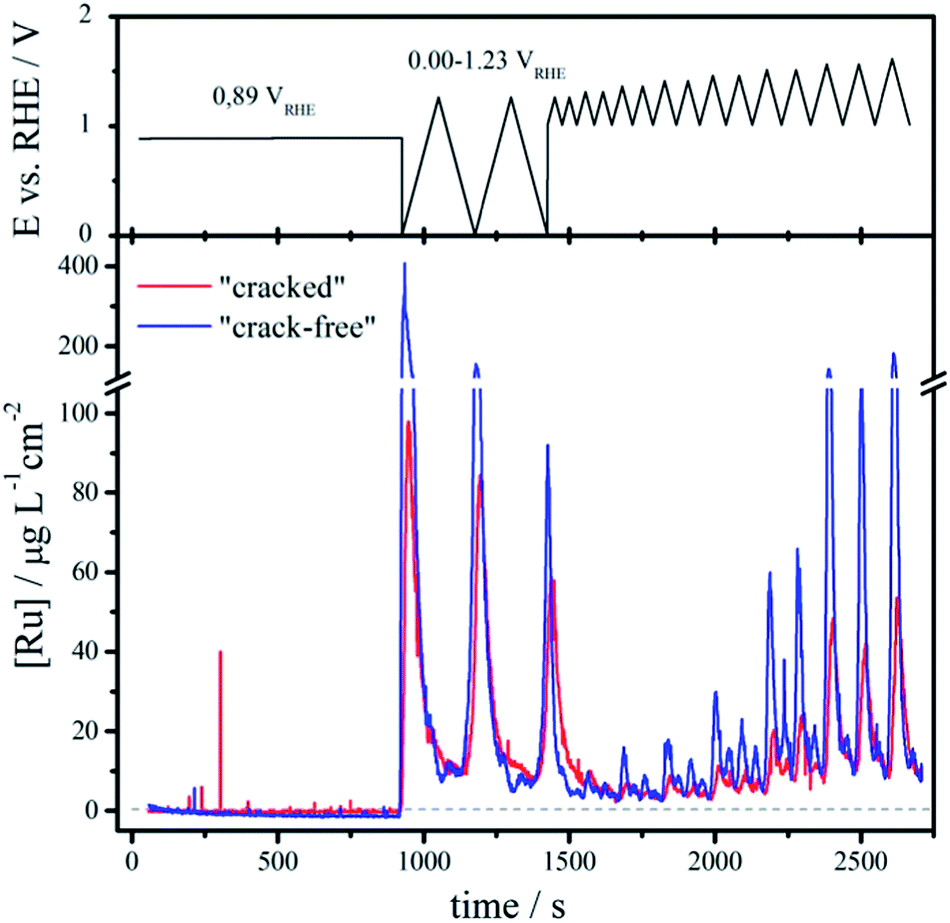 | ||
| Fig. 4 Normalized dissolution profile of Ru to the geometric area for the two samples with similar chemical composition and loading, but different morphological pattern: “cracked” coating (red) and “crack-free” coating (blue). The applied potential profile is shown on top. The sweep rate in the cyclic voltammograms was 10 mV s−1. The dashed line indicates the detection limit. | ||
Initially, the samples are kept at open circuit conditions for around 900 s (OCP at ca. 0.89 VRHE). Then the potential is cycled in the region of thermodynamic stability of water (0.00–1.23 VRHE) where particularly the first cathodic sweep from OCP to 0.00 VRHE causes massive dissolution. The onset of dissolution is estimated around 0.50 VRHE, which corresponds to the redox transition from RuO2(c) to Ru3+.50 While such low potentials do not typically occur during oxygen evolution, still they can be induced in case of cell shutdown due to hydrogen cross-over. The integrated area beneath the initial dissolution peak (range 0.00–1.23 VRHE) is around 43 ng cm−2 and 21 ng cm−2 for the “crack-free” and the “cracked” sample, respectively. With continuous potential cycling the amount of dissolved Ru decreases and approaches similar values, so that in the third cycle they are as low as 13 ng cm−2 and 12 ng cm−2. According to this it seems that the cathodic dissolution strongly depends on the prehistory of the samples, particularly on the oxide coverage at OCP.
Afterwards the potential is cycled between a fixed negative potential limit of approximately 1.00 VRHE and a varying positive limit (from 1.25 to 1.60 VRHE with increment of 50 mV for every second cycle). The dissolution in this case appears at positive potentials above approximately 1.25 VRHE, which coincides with the reversible potential of the OER. Notice that the dissolution is always higher for the “crack free” sample than the “cracked” one. With increasing the upper potential limit the dissolution gradually increases and becomes severe above 1.45 VRHE, which corresponds to the redox transition between RuO2(c) and RuO4, 4H+.50 Ruthenium tetroxide formed during the OER has previously been identified in situ by Raman spectroscopy51 and is known to be unstable/soluble in water.52 Moreover, studies based on differential electrochemical spectroscopy using isotope labeling53 substantiate that the OER on Ru based electrodes proceeds through a surface oxide route, opposite to some other cases as for example on platinum based electrodes.54 Thus, also the disruption of the surface oxide during the OER on Ru-based electrodes can cause enhanced dissolution at positive potentials.25,53,55 Without a clear notion if the formation of RuO4 complemented by the disruption of the surface during the OER is essential for dissolution, it can be concluded that any activity improvement that can shift the operation potential below a critical value for anodic dissolution (around 1.45 VRHE), will have multiple implications; namely (i) reduction of the electrical energy consumption, (ii) inhibition of Ru dissolution, and lastly (iii) the reaction would become endothermic due to the fact that the thermoneutral voltage is around 1.48 VSHE.56 In Table 2 the most important conclusions for a conceptual understanding of the process of Ru dissolution from RuO2 are summarized.
| Potential range, vs. RHE/V | Type of dissolution | Onset of dissolution vs. RHE/V | Coincident redox transition |
|---|---|---|---|
| 0.00–1.23 | Negative scan | 0.50 | RuO2(c), H+/Ru3+ |
| 1.00–1.60 | Positive potentials | 1.23 | H2O/O2, 4H+ |
4.4. The impact of the surface morphology on the catalyst performance
Relating the rate of dissolution with the rate of OER57 it seems that for the two analyzed samples with similar chemistry the turnover per available active site has to be very similar. Considering that Ru dissolution of the cracked sample is half compared to the crack-free sample and that the current intensity for the cracked sample is two times higher, the effective surface area of the cracked sample is approximately four times higher. This suggests that in the case of the cracked sample not only inner surface is utilized, but also its outer surface participates more frequently in the reaction. The gas-bubble behavior of both samples is additionally studied to gain better insight into this effect.As it has been shown already for other similar reactions like the chlorine evolution (CER), the evolution of gas-bubbles from the surface is indeed one of the main contributors to the macrokinetics. As a consequence a difference between the two types of coatings should be reflected also in different rates for gas-bubble removal. The latter can be estimated from an experimental approach based on monitoring of the pseudo-oscillatory behavior of gas evolution using SECM.22 A periodical release of a gas fraction from the surface causes local changes in the ohmic resistance of the electrolyte between a positioned microelectrode tip and a reference electrode. The recorded temporal fluctuations of the current at the microelectrode tip can then be processed using Fast Fourier analysis. The frequency of the gas-bubble detachment can thus be monitored as a function of the applied anode potential.
The characteristic gas-bubble detachment during OER as estimated from SECM is presented in the form of potential dependent frequency spectra in Fig. 5. The two different morphological patterns exhibit markedly different frequency spectra. In the case of the “cracked” surface a definite frequency of gas-bubble detachment can be extracted, which increases with the applied electrode potential (Fig. 5a). The initial potential of gas-bubble detachment is around 1.7 VRHE (without ohmic drop correction), and the frequency increases from 2 Hz towards 4 Hz in the potential range between 1.7 and 2.0 VRHE. In contrast, no characteristic frequency could be detected in the case of “crack-free” electrodes, and the spectra in Fig. 5b imply only chaotic gas bubble detachment without any recognizable pattern. A similar behavior was observed previously for the CER, where only certain morphological patterns exhibited defined frequencies for gas-bubble detachment.22 An uniform oscillatory behavior of the system was strongly related with higher activities and certain morphological features in this case, as it is here for the OER. Comparing the absolute values of normalized activity (1.55 Hz at 1.45 VRHE) and frequency of gas-bubble detachment (2 Hz at 1.7 VRHE), it seems that the removal of gas-bubbles markedly influences macrokinetics of OER and consequently the almost two times higher activity of the “cracked” sample could originate from the faster and more regular removal of the gas-bubbles from the “outer” surface. This establishes a highly frequent contact between the electrolyte and the solid surface, and thus significantly extends the average availability of the active outer surface sites.
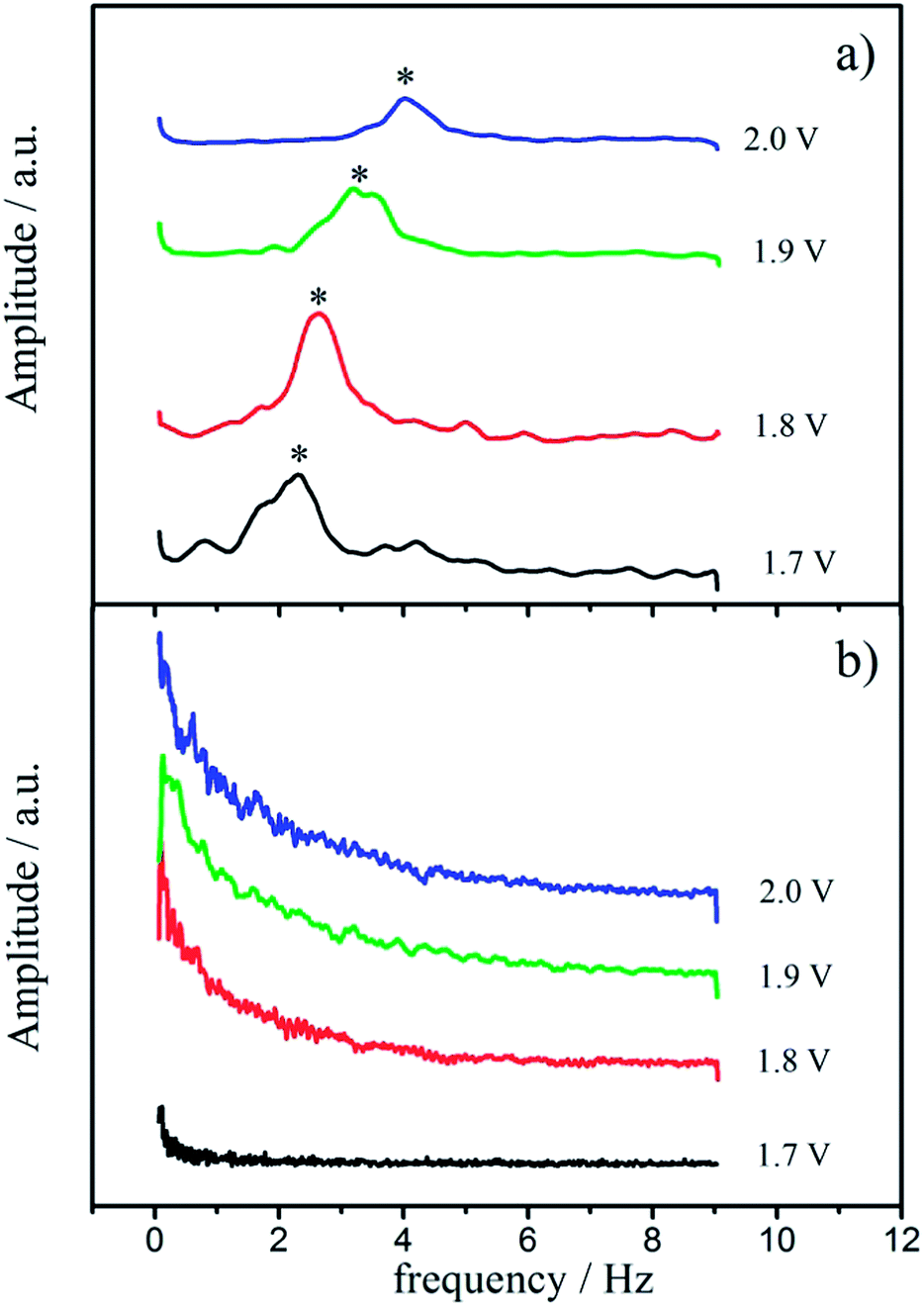 | ||
| Fig. 5 Potential dependent frequency spectra of the (a) “cracked” and the (b) “crack-free” RuO2 based electrodes, as measured via SECM. Potentials are given with respect to RHE and without ohmic drop correction (reference electrode however always at the same distance from the working electrode). | ||
To comprehend why “cracked” electrodes exhibit more facile gas-bubble removal than the “crack-free” electrodes it is important to consider that gas evolution during OER can be described with three processes: (1) nucleation of gas-bubbles (2) bubble growth and coalescence and (3) bubble detachment.
The radius of gas-bubble nucleation in typical acidic electrolyte is around 1 μm at 25 °C and 1 bar.58 Thus, when the crack width and the thickness of the catalyst coating allow gas-bubble nucleation inside the cracks, their growth will be limited in contrast to an open, crack-free surface (see scheme in Fig. 6). Furthermore, during the growth phase the bubbles are in constant oscillatory motion due to thermal fluctuations (similar to boiling) and cause an oscillatory motion of the surrounding liquid. This in turn the root of the bubble and its contact with the hydrophilic surface, what finally causes the bubble to detach. The oscillations of smaller bubbles are more frequent than in the case of larger bubbles, so that the detachment probability of bubbles limited in size within cracks is higher. This is in line with the theoretically established inverse proportionality between the radius of gas-bubble and frequency of gas-bubble detachment,59 which suggests that particularly small bubbles at the solid/liquid interface induce efficient gas evolution. Certainly, the growth of gas-bubbles at “terraces” is also possible in the case of “cracked” electrodes, but it will be limited by the spacing between the cracks and frequent microconvective flow of electrolyte towards the “cracks”. These considerations suggest that controlling the size of the “cracks”, their width and the thickness of the catalyst coating is a central task for designing efficient electrode for OER.
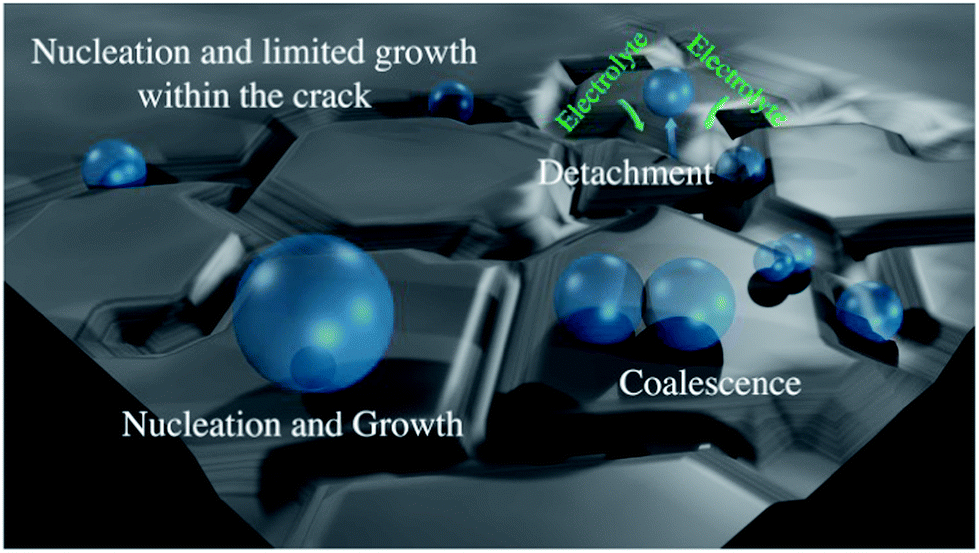 | ||
| Fig. 6 Illustration of the gas-bubble behavior at the “outer” surface of the “cracked” electrode. | ||
5. Conclusions
The design of the surface morphology of the anode is shown to be a very promising strategy to improve the efficiency of the oxygen evolution reaction and thus the overall energy savings during electrochemical fuel production. Simultaneous gain in catalyst activity and stability was shown to be realistic consequence of facile removal of gas-bubbles from the electrode surface. It was essential to create morphological patterns that promote nucleation of gas-bubbles with limited growth in confined regions and that cause high detachment frequencies. It was shown that the design of morphological patterns has a rational basis in the control of the tensile stress in the catalyst coating during the synthesis. Intentionally induced cracks in the surface with well controlled spacing can indeed enhance the bubble detachment, and thus improve the catalyst performance.Acknowledgements
BMBF for the financial support in the framework of the project ECCO2 (Kz: 033RC1101A) as well as the Helmholtz-Energie-Allianz “Stationäre elektrochemische Speicher und Wandler” (HA-E-0002) and the Cluster of Excellence RESOLV (DFG EXC-1069).References
- M. Redclift, Global Environ. Change, 1992, 2, 32–42 CrossRef.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933 RSC.
- J. Greeley and N. M. Markovic, Energy Environ. Sci., 2012, 5, 9246 CAS.
- K. J. P. Schouten, Y. Kwon, C. J. M. van der Ham, Z. Qin and M. T. M. Koper, Chem. Sci., 2011, 2, 1902 RSC.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 2000, 45, 2377–2385 CrossRef CAS.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Ind. Ed., 2012, 51, 12613–12615 CrossRef PubMed.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. T. M. Koper, J. Electroanal. Chem., 2011, 660, 254–260 CrossRef CAS PubMed.
- J. O. Bockris, J. Electrochem. Soc., 1984, 131, 290 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- M. García-Mota, M. Bajdich, V. Viswanathan, A. Vojvodic, A. T. Bell and J. K. Nørskov, J. Phys. Chem. C, 2012, 116, 21077–21082 Search PubMed.
- G. N. Martelli, R. Ornelas and G. Faita, Electrochim. Acta, 1994, 39, 1551–1558 CrossRef CAS.
- H. Vogt, J. Appl. Electrochem., 1993, 23, 1323–1325 CrossRef CAS.
- H. Vogt, Electrochim. Acta, 2012, 78, 183–187 CrossRef CAS PubMed.
- H. Vogt, Electrochim. Acta, 1984, 29, 167–173 CrossRef CAS.
- A. R. Zeradjanin, F. La Mantia, J. Masa and W. Schuhmann, Electrochim. Acta, 2012, 82, 408–414 CrossRef CAS PubMed.
- V. Trieu, B. Schley, H. Natter, J. Kintrup, A. Bulan and R. Hempelmann, Electrochim. Acta, 2012, 78, 188–194 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- A. R. Zeradjanin, E. Ventosa, A. S. Bondarenko and W. Schuhmann, ChemSusChem, 2012, 5, 1905–1911 CrossRef CAS PubMed.
- A. R. Zeradjanin, T. Schilling, S. Seisel, M. Bron and W. Schuhmann, Anal. Chem., 2011, 83, 7645–7650 CrossRef CAS PubMed.
- A. A. Topalov, I. Katsounaros, M. Auinger, S. Cherevko, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2012, 51, 12613–12615 CrossRef CAS PubMed.
- S. Cherevko, A. A. Topalov, A. A. Zeradjanin, I. Katsounaros and K. J. J. Mayrhofer, RSC Adv., 2013, 3, 16516–16527 RSC.
- K. Kim, J. Catal., 1974, 35, 66–72 CrossRef CAS.
- S. O. Klemm, A. A. Topalov, C. A. Laska and K. J. J. Mayrhofer, Electrochem. Commun., 2011, 13, 1533–1535 CrossRef CAS PubMed.
- A. A. Topalov, I. Katsounaros, J. C. Meier, S. O. Klemm and K. J. J. Mayrhofer, Rev. Sci. Instrum., 2011, 82, 114103 CrossRef PubMed.
- H. Over, Electrochim. Acta, 2013, 93, 314–333 CrossRef CAS PubMed.
- J. Lee, B. Jeong and J. D. Ocon, Curr. Appl. Phys., 2013, 13, 309–321 CrossRef PubMed.
- R. Chen, V. Trieu, A. R. Zeradjanin, H. Natter, D. Teschner, J. Kintrup, A. Bulan, W. Schuhmann and R. Hempelmann, Phys. Chem. Chem. Phys., 2012, 14, 7392 RSC.
- J. W. Hutchinson and T. Y.-T. Wu, Advances in applied mechanics, Academic Press, New York, 1991, vol. 29 Search PubMed.
- F. Delannay and P. Warren, Acta Metall. Mater., 1991, 39, 1061–1072 CrossRef CAS.
- R. D. Shannon, Solid State Commun., 1968, 6, 139–143 CrossRef CAS.
- J. Gaudet, A. C. Tavares, S. Trasatti and D. Guay, Chem. Mater., 2005, 17, 1570–1579 CrossRef CAS.
- R. Kötz and S. Stucki, Electrochim. Acta, 1986, 31, 1311–1316 CrossRef.
- R. Chen, V. Trieu, B. Schley, H. Natter, J. Kintrup, A. Bulan, R. Weber and R. Hempelmann, Z. Phys. Chem., 2013, 227, 651–666 CrossRef CAS.
- C. Angelinetta, S. Trasatti, L. D. Atanososka and R. T. Atanasoski, J. Electroanal. Chem. Interfacial Electrochem., 1986, 214, 535–546 CrossRef CAS.
- L. D. Burke, O. J. Murphy and J. F. O'Neill, J. Electroanal. Chem. Interfacial Electrochem., 1977, 81, 391–394 CrossRef CAS.
- L. D. Burke and O. J. Murphy, J. Electroanal. Chem. Interfacial Electrochem., 1979, 96, 19–27 CrossRef CAS.
- L. D. Burke and O. J. Murphy, J. Electroanal. Chem. Interfacial Electrochem., 1980, 112, 39–50 CrossRef CAS.
- S. Ardizzone, J. Electrochem. Soc., 1982, 129, 1689 CrossRef CAS PubMed.
- S. Ardizzone, G. Fregonara and S. Trasatti, Electrochim. Acta, 1990, 35, 263–267 CrossRef CAS.
- D. R. Rolison, Science, 2003, 299, 1698–1701 CrossRef CAS PubMed.
- S. V. Evdokimov, Russ. J. Electrochem., 2000, 36, 236–239 CrossRef CAS.
- S. V. Evdokimov, Russ. J. Electrochem., 2000, 36, 489–494 CrossRef CAS.
- S. Trasatti and O. A. Petrii, J. Electroanal. Chem., 1992, 327, 353–376 CrossRef CAS.
- R. F. Savinell, J. Electrochem. Soc., 1990, 137, 489 CrossRef CAS PubMed.
- E. H. Calderon, A. Katsaounis, R. Wüthrich, P. Mandin, G. Foti and C. Comninellis, J. Appl. Electrochem., 2009, 39, 1827–1833 CrossRef CAS PubMed.
- S. G. Bratsch, J. Phys. Chem. Ref. Data, 1989, 18, 1 CrossRef CAS PubMed.
- A. R. Zeradjanin, N. Menzel, P. Strasser and W. Schuhmann, ChemSusChem, 2012, 5, 1897–1904 CrossRef CAS PubMed.
- P. Patnaik, Handbook of inorganic chemicals, McGraw-Hill, New York, 2003 Search PubMed.
- M. Wohlfahrt-Mehrens and J. Heitbaum, J. Electroanal. Chem. Interfacial Electrochem., 1987, 237, 251–260 CrossRef CAS.
- J. Willsau, O. Wolter and J. Heitbaum, J. Electroanal. Chem. Interfacial Electrochem., 1985, 195, 299–306 CrossRef CAS.
- O. Diaz-Morales, F. Calle-Vallejo, C. de Munck and M. T. M. Koper, Chem. Sci., 2013, 4, 2334 RSC.
- K. Zeng and D. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS PubMed.
- B. V. Tilak, V. I. Birss, J. Wang, C.-P. Chen and S. K. Rangarajan, J. Electrochem. Soc., 2001, 148, D112 CrossRef CAS PubMed.
- A. G. Pshenichnikov, Y. G. Chirkov and V. I. Rostokin, Russ. J. Electrochem., 2002, 38, 213–219 CrossRef CAS.
- I. G. Malenkov, J. Eng. Phys., 1971, 20, 704–708 CrossRef.
| This journal is © The Royal Society of Chemistry 2014 |