
Pure gold nanocages by galvanic replacement reaction of magnesium nanoparticles†
Abstract
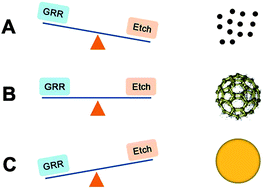
We develop an integrative-gas-liquid strategy to produce Au nanocages with high purity, where Mg nanoparticles are first generated by laser ablation and then blown into aqueous solution for growing Au nanostructures. The Au nanocages exhibit a high DOX-loading capacity, which favors biomedical applications.
 Please wait while we load your content...
Please wait while we load your content...

