
Enhanced luminescence and charge separation in polythiophene-grafted, gold nanoparticle-decorated, 1-D ZnO nanorods
Abstract
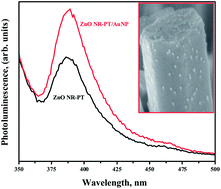
In this work, we demonstrate a facile method for fabricating hybrid organic/inorganic structures of vertically oriented 1-dimensional (1-D) ZnO nanorods (NR) coupled with regio-regular poly(3-(2-methoxyethoxy) ethoxymethyl) thiophene-2,5-diyl (PMEEMT). The NRs were synthesized by two-step solution processes and exhibited a hexagonal wurtzite structure with a lattice spacing of 0.248 nm. Cross-section analysis of field-emission scanning electron microscopy (FESEM) analysis confirmed the NRs were vertically oriented and densely packed with diameter of about 250 nm. The hybrid structures were formed by encapsulating the ZnO NRs with polythiophenes, thereby creating a direct heterojunction interface. These interfaces allowed easy electron transport along the NR avoiding loss of mobility due to grain boundary scattering; further, the large aspect ratio and surface area of NR increased the effective interface between polythiophene and the nanorods contributing to efficient charge separation. In addition, decoration of the ZnO hybrid structures by gold nanoparticles (AuNPs) induced enhanced luminescence properties. These functionalized organic/inorganic hybrid structures can pave the way for applications in photovoltaics and sensing.
 Please wait while we load your content...
Please wait while we load your content...

