
Validity and limitations of the annulene-within-an-annulene (AWA) model for macrocyclic π-systems
Abstract
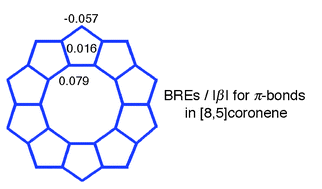
Recently some macrocyclic π-systems have been proposed as possible candidates for the annulene-within-an-annulene (AWA) model. We found that bond resonance energies (BREs) for spokes that link outer and inner annulenic subsystems can be used as an excellent measure for deciding whether or not a given macrocycle conforms to the AWA picture. In general, macrocycles with small BRE(spoke) values, such as [10,5]coronene and [14,7]coronene, can be described well in terms of the AWA model.
 Please wait while we load your content...
Please wait while we load your content...

