
Selective “turn-off” fluorescent sensing of mercury ions using aminocyclodextrin:3-hydroxy-N-phenyl-2-naphthamide complex in aqueous solution†
Abstract
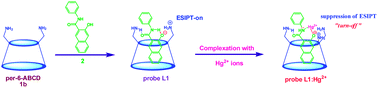
A sensitive and highly selective, fluorescent “turn-off” colorimetric sensor for Hg2+ ions is reported using an aminocyclodextrin:3-hydroxy-N-phenyl-2-naphthamide complex in a 5% CH3CN–water system. Six different aminocyclodextrins viz., per-6-amino-α-cyclodextrin, per-6-amino-β-cyclodextrin, per-6-amino-γ-cyclodextrin, mono-6-amino-α-cyclodextrin, mono-6-amino-β-cyclodextrin and mono-6-amino-γ-cyclodextrin were synthesized and used as hosts for complexing 3-hydroxy-N-phenyl-2-naphthamide. This complex can be used as a sensing system for Hg2+ ions. A blue shifted “turn-off” fluorescence quenching and color change from yellow to colorless in the presence of Hg2+ ions is observed which is attributed to suppression of excited-state intramolecular processes (ESIPT) upon Hg2+ ion complexation. Selectivity towards Hg2+ is found to depend upon cavity size/close proximity of the amino groups of aminocyclodextrins (L1–L6) to the metal ion. Probe L1 is selective and sensitive to Hg2+ with a detection limit as low as 1 × 10−12 M. The present sensor system can also be applied to detect the level of Hg2+ in real environmental water samples.
 Please wait while we load your content...
Please wait while we load your content...

