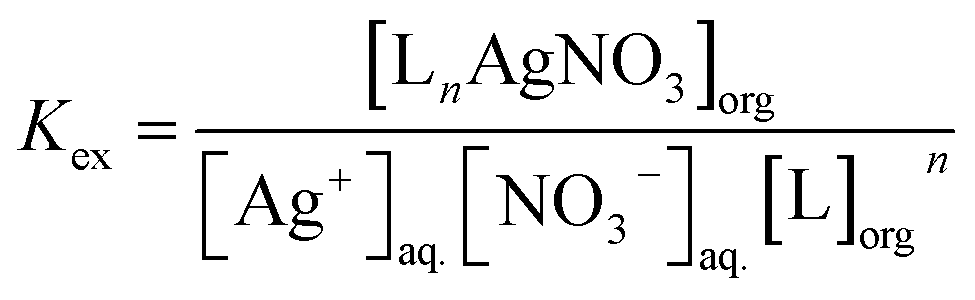DOI:
10.1039/C3RA45700A
(Paper)
RSC Adv., 2014,
4, 9791-9798
Steric hindrance effects in tripodal ligands for extraction and back-extraction of Ag+†
Received
10th October 2013
, Accepted 27th January 2014
First published on 28th January 2014
Abstract
A novel series of tripodal ligands with thiophenylether arms connected to an anchoring nitrogen has been investigated. Seven tripodal ligands were synthesized by combining methyl, isopropyl, and tert-butyl residue bearing thiophenylether sites as groups with different steric hindrance effects. The tripodal ligands allowed for the extraction of Ag+ ions from the aqueous phase into a chloroform phase by forming 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complexes with Ag+. Back-extraction was performed with 1 M HNO3 aqueous solution. Each ligand showed different extraction and back-extraction efficiency for Ag+, affected by changes in steric hindrance caused by the various combinations of sidearms. These results are further supported by X-ray single crystal structural analysis.
:1 complexes with Ag+. Back-extraction was performed with 1 M HNO3 aqueous solution. Each ligand showed different extraction and back-extraction efficiency for Ag+, affected by changes in steric hindrance caused by the various combinations of sidearms. These results are further supported by X-ray single crystal structural analysis.
Introduction
Coordination chemistry is relevant for complex formation and molecular recognition. In this context, tripodal ligands having multidentate structure have attracted special interest. A tripodal ligand is characterized by its molecular structure with three coordinating arms. Typically, each of the coordinating arms having one or more donor atoms (N, O, S, P) is connected to an anchoring nitrogen or phosphorous atom.1 As coordinating arms, pyridyl,2 quinolyl,3 alkoxyl,4 and thioether5 groups have been presented, among others. In particular, tripodal ligands have been used as extractants for metals.6–10
From an economic and environmental standpoint, it is of great importance to recover expensive metals for reuse and recycling from electronic waste, or to remove metal contaminants from wastewater.11–15 Examples include heavy metals, radioactive precious metals, rare metals, and rare earth metals. Solvent extraction is a very useful method to separate and concentrate metal cations in solution, and is widely applied for separations on industrial scale, such as in hydrometallurgy. There are many reports on the extraction of metal cations using various kinds of extractants, including studies focusing on the relationship between extraction efficiency and ligand structure.16–18
On the other hand, studies focusing on the relationship between back-extraction efficiency and ligand structure are hardly found, so far. In the back-extraction process, the target component is transferred back to the aqueous phase by extraction of the organic phase with an aqueous solution under conditions shifting the distribution equilibrium of the target component away from the organic phase. That distribution equilibrium is controlled by the reagent concentrations, and the pH of the aqueous phase. In a metal recycling process, the back-extraction represents a second extraction step, and allows for further selectivity improvement by choosing a suitable aqueous phase composition. For example, Narita et al. successfully recovered Rh prior to Pd and Pt by effective control of back-extraction conditions.19
According to the HSAB concept,20 ‘soft’ acids such as transition metal ions, and sulfur atoms characterized as ‘soft’ bases generally have a strong affinity for each other. Therefore, there are a variety of sulfur containing structures reported as ligands for soft metal ions, in the form of macrocycles,21 crown ethers,22 lariat ethers,23 cryptands,24 calixarenes,25 and tripodal ligands.26 However, with a strong coordination bond formed between the soft sulfur donor site and a soft transition metal ion like Ag+, the use of an aqueous mineral acid is no longer sufficient for the efficient release of the metal cation for back-extraction. In that case, compounds with stronger interaction with Ag+, such as ammonia or thiourea, are required in the aqueous phase.27
In the present work, the influence of steric hindrance in a tripodal ligand on the back-extraction efficiency with an aqueous mineral acid was investigated. For this purpose, a new series of tripodal ligands (6a–g) with three thiophenylether sidearms connected to an anchoring nitrogen has been synthesized and characterized. The effect of steric hindrance induced by combinations of methyl, isopropyl, and tert-butyl substituted thiophenylether sidearms on extraction and back-extraction efficiencies of Ag+ was evaluated. In addition, X-ray crystal structure analyses of the tripodal ligands with Ag+ have been performed for studying the binding structure.
Experimental
Reagents and instruments
All reagents for the syntheses of tripodal ligands (6a–g) were purchased from the following commercial suppliers and were used without further purification: Wako Pure Chemical (Osaka, Japan), Tokyo Kasei Industry (Tokyo, Japan) and Aldrich Chemical (St. Louis, MO, U.S.A.). 1H-NMR and 13C-NMR spectra were recorded at room temperature on a JEOL ECA-500 spectrometer at 500 MHz and 125 MHz, respectively. All chemical shifts are relative to an internal standard of tetramethylsilane (δ = 0.0 ppm), and coupling constants are given in Hz. High-resolution mass spectra were obtained on a Waters Xevo G2-S QTof MS. ESI-mass spectra were obtained on a LCMS-2010EV (SHIMADZU, Kyoto, Japan). Flash chromatography separation was undertaken using a YFLC-Al-560 chromatograph (Yamazen Co., Osaka, Japan). ICPS-8000 inductively coupled argon plasma atomic emission spectrometry (SHIMADZU, Kyoto, Japan) was used for the determination of the concentration of each metal ion in aqueous solution. The deionized water used had a resistivity of 18.2 MΩ at 25 °C. The pH values were verified using an IOL-50 ion-meter (DKK-TOA Corp., Tokyo, Japan) with a glass pH electrode. All X-ray crystal structure measurements were performed with a Rigaku Saturn70 diffractometer using multi-layer mirror monochromated Mo-Kα (λ = 0.71075 Å) radiation. The structure was solved by direct methods, and expanded using Fourier techniques. Non-hydrogen atoms were refined anisotropically. Structural refinements were obtained with full-matrix least-squares based on F2 using the program SHELXL-97.28
Synthesis
2-Thio-substituted benzoic acid (2a and 2b) was prepared as previously reported.29
2-(tert-Butylthio)benzoic acid (2c). 1 (4.92 g, 31.9 mmol) was dissolved in a mixture of acetic acid (46 mL), 60 % perchloric acid (10 mL), acetic anhydride (8 mL) and THF (40 mL). Then, tertiary butyl alcohol (4.68 g, 63.1 mmol) was added and the solution was stirred for 18 h at room temperature, followed by 2 h at 50 °C. After the reaction was completed, the reaction mixture was allowed to cool to room temperature, and excess saturated aqueous NaHCO3 solution was added to hydrolyze remaining acetic anhydride. After that, the pH of the solution was lowered to pH 1 by addition of 1 M HCl solution. The acidic aqueous phase was extracted with ethyl acetate 3 times, and the combined organic layer was washed with water two times and brine once, dried over Na2SO4, and the solvent evaporated. The resulting residue was used for the following reaction without further purification.
General procedure for the synthesis of 2-thio-substituted phenylmethanol compounds (3a–c). The corresponding 2-thio-substituted benzoic acid 2a–c was dissolved in THF (200 mL) and then cooled in an ice-bath (0 °C). 1 M BH3·THF (70 mL) was added to the flask, and the reaction was stirred at 0 °C for 1 h and then at room temperature for 3 h. It was then cooled in an ice bath and quenched by the careful addition of water. Solid K2CO3 was added until the solution was saturated, and the layers were separated. The organic layer was evaporated to dryness, while the aqueous phase was extracted with diethylether 2 times. All of the organic phases were combined, washed with 1 M NaOH, water, and brine, dried over Na2SO4, evaporated, and dried under vacuum.
(2-(Methylthio)phenyl)methanol (3a). Compound 2a (14.1 g, 83.7 mmol) was used as the starting material, and 3a was obtained as clear oily liquid (12.2 g, 94.3%). 1H-NMR (CDCl3, 500 MHz) δ (ppm): 2.49 (s, 3H), 4.75 (s, 2H), 7.18–7.20 (m, 1H), 7.26–7.29 (m, 2H), 7.37 (d, J = 7.4 Hz, 1H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 16.21, 63.59, 125.61, 126.68, 128.11, 128.49, 136.76, 138.98.
(2-(Isopropylthio)phenyl)methanol (3b). Compound 2b (4.98 g, 25.4 mmol) was used as the starting material, and 3b was obtained as light yellow oily liquid (4.54 g, 98.2%). 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.30 (d, J = 6.9 Hz, 6H), 2.47 (br, 1H), 3.38 (sep, J = 6.9 Hz, 1H), 4.79 (s, 2H), 7.24–7.25 (m, 2H), 7.39–7.45 (m, 2H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 23.30, 38.97, 64.11, 127.48, 128.21, 128.57, 133.04, 133.94, 142.40.
(2-(tert-Butylthio)phenyl)methanol (3c). Compound 2c was used as the starting material, and 3c was obtained as light yellow oily liquid (5.81 g, 93.0%). 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.31 (s, 9H), 2.52 (br, 1H), 4.87 (s, 2H), 7.25–7.28 (m, 1H), 7.35–7.38 (m, 1H), 7.47 (d, J = 7.5 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 31.30, 47.76, 64.67, 127.70, 128.75, 129.45, 131.15, 138.96, 145.94.Alkyl-substituted (2-(bromomethyl)phenyl)sulfates (4a and 4b) were prepared as previously reported.29
(2-(Bromomethyl)phenyl)(tert-butyl)sulfate (4c). Compound 3c (5.81 g, 29.6 mmol) was dissolved in diethylether (300 mL). After cooling to 0 °C, PBr3 (4.00 g, 14.8 mmol) was added and the reaction was stirred at 0 °C for 0.5 h and room temperature for 1 h. After the reaction was completed, the reaction was quenched with methanol. Then, water was added, and the organic layer was washed with saturated NaHCO3 three times and dried over Na2SO4, evaporated and dried under vacuum. 4c was obtained as colorless oily liquid (6.70 g, 87.4%).1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.32 (s, 9H), 4.88 (s, 2H), 7.24–7.27 (m, 1H), 7.33–7.36 (m, 1H), 7.56 (d, J = 7.7 Hz, 2H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 31.38, 32.80, 47.47, 128.47, 129.60, 131.13, 132.62, 139.02, 142.96.
General procedure for the synthesis of 2-thio-substituted phenylmethanamines (5a–c). The corresponding alkyl-substituted (2-(bromomethyl)phenyl)sulfate 4a–c was dissolved in ethanol (160 mL). Then, 28–30 wt% aqueous ammonia solution (80 mL) and THF (80 mL) were added, and the reaction was stirred at room temperature overnight. After the reaction was completed, the solvent was evaporated and the residue was dried under vacuum, before being purified by flash chromatography (silica gel).
(2-(Methylthio)phenyl)methanamine (5a). Compound 4a (3.61 g, 16.6 mmol) was used as the starting material, and 5a was obtained as a white powder (2.20 g, 86.4%). Eluent for chromatography: chloroform–methanol 94:6 → 90:10 → 85:15. 1H-NMR (CD3OD, 500 MHz) δ (ppm): 2.54 (s, 3H), 4.24 (s, 2H), 7.26–7.28 (m, 1H), 7.41–7.47 (m, 3H). 13C-NMR (CD3OD, 125 MHz) δ (ppm): 16.53, 41.98, 127.17, 128.97, 130.83, 131.19, 132.74, 139.62.
(2-(Isopropylthio)phenyl)methanamine (5b). Compound 4b (4.51 g, 18.4 mmol) was used as the starting material, and 5b was obtained as a white powder (2.68 g, 80.4%). Eluent for chromatography: chloroform–methanol 100:0 → 98:2 → 50:50. 1H-NMR (CD3OD, 500 MHz) δ (ppm): 1.29 (d, J = 6.9 Hz, 6H), 3.41 (sep, J = 6.9 Hz, 1H), 4.34 (s, 2H), 7.37–7.44 (m, 2H), 7.52 (d, J = 7.5 Hz, 1H), 7.61 (dd, J = 7.5 Hz, 1.2 Hz, 1H). 13C-NMR (125 MHz, CD3OD) δ (ppm): 23.36, 40.58, 42.56, 129.23, 130.91, 131.03, 135.25, 136.27, 136.68.
(2-(tert-Butylthio)phenyl)methanamine (5c). Compound 4c (2.31 g, 8.92 mmol) was used as the starting material, and 5c was obtained as a white powder (1.30 g, 69.3%). Eluent for chromatography: chloroform–methanol 90:10. 1H-NMR (CD3OD, 500 MHz) δ (ppm): 1.30 (s, 9H), 4.47 (s, 2H), 7.44–7.47 (m, 1H), 7.50–7.53 (m, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.69 (d, J = 7.8 Hz, 1H). 13C-NMR (CD3OD, 125 MHz) δ (ppm): 30.29, 42.19, 129.54, 130.22, 130.25, 133.19, 138.58, 139.69.
General procedure for the synthesis of alkyl-substituted N,N-bis(2-thiobenzyl)-1-(2-thiophenyl)-methanamines (6a–g). The corresponding alkyl-substituted (2-(bromomethyl)phenyl)sulfate (4a–c), 2-thio-substituted phenylmethanamine (5a–c) and K2CO3 were dissolved in acetonitrile and then stirred at room temperature for 1 day. After the reaction was completed, solids were removed by filtration, and the solvent was removed by evaporation. The resulting residue was purified by flash chromatography (silica gel).
Tris(2-(methylthio)benzyl)amine (6a). Compound 4a (1443 mg, 6.64 mmol), compound 5a (506 mg, 3.30 mmol) and K2CO3 (2.00 g, 14.5 mmol) in acetonitrile (200 mL) were used. 6a was obtained as a white powder (1024 mg, 72.4%). Eluent for chromatography: chloroform–methanol 100:0 → 99:1 → 90:10. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 2.40 (s, 9H), 3.74 (s, 6H), 7.13–7.25 (m, 9H), 7.69 (d, J = 7.5 Hz, 3H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 16.13, 55.79, 125.05, 125.54, 127.31, 128.84, 137.55, 137.67. HRMS: calculated for C24H27NS3 426.1384 [M + H]+; found 426.1387. Elemental analysis: calcd C 67.72, H 6.39, N 3.29, S 22.60; found C 67.73, H 6.55, N 3.22, S 22.61.
N-(2-(Isopropylthio)benzyl)-N-(2-(methylthio)benzyl)-1-(2-(methylthio)phenyl)methanamine (6b). Compound 4a (1180 mg, 5.43 mmol), compound 5b (504 mg, 2.78 mmol) and K2CO3 (2.00 g, 14.5 mmol) in acetonitrile (250 mL) were used. 6b was obtained as lightly yellow solid (773 mg, 61.2%). Eluent for chromatography: hexane–ethyl acetate 88:12 → 80:20 → 50:50. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.25 (d, J = 6.6 Hz, 6H), 2.41 (s, 6H), 3.29 (sep, J = 6.6 Hz, 1H), 3.73 (s, 4H), 3.84 (s, 2H), 7.10–7.22 (m, 8H), 7.36 (dd, J = 7.8 Hz, 1.2 Hz, 1H), 7.68 (d, J = 7.2 Hz, 2H), 7.76 (dd, J = 7.5 Hz, 1.2 Hz, 1H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 16.09, 23.24, 38.35, 55.81, 56.06, 125.00, 125.50, 126.85, 126.98, 127.28, 128.84, 129.33, 131.96, 135.08, 137.61, 137.69, 140.90. HRMS: calculated for C26H32NS3 454.1652 [M + H]+; found 454.1701. Elemental analysis: calcd C 68.83, H 6.89, N 3.09, S 21.20; found C 68.77, H 6.96, N 2.94, S 21.36.
N,N-Bis(2-(isopropylthio)benzyl)-1-(2-(methylthio)phenyl)-methanamine (6c). Compound 4b (2240 mg, 9.13 mmol), compound 5a (700 mg, 4.57 mmol) and K2CO3 (2.83 g, 20.5 mmol) in acetonitrile (330 mL) resulted in 6c as lightly yellow oily liquid (1500 mg, 68.3%). Eluent for chromatography: hexane–chloroform 75:25 → 20:80 → 0:100. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.24 (d, J = 6.9 Hz, 12H), 2.40 (s, 3H), 3.28 (sep, J = 6.9 Hz, 2H), 3.72 (s, 2H), 3.83 (s, 4H), 7.12–7.21 (m, 7H), 7.36 (dd, J = 7.7 Hz, 1.6 Hz 2H), 7.67 (d, J = 7.2 Hz, 1H), 7.76 (dd, J = 7.7 Hz, 1.2 Hz, 2H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 16.05, 23.23, 38.32, 55.81, 56.05, 124.95, 125.44, 126.83, 126.95, 127.25, 128.84, 129.35, 131.96, 135.10, 137.67, 137.72, 141.00. HRMS: calculated for C28H35NS3 482.1965 [M + H]+; found 482.2012. Elemental analysis: calcd C 69.80, H 7.32, N 2.91, S 19.97; found C 69.71, H 7.47, N 2.79, S 20.41.
Tris(2-(isopropylthio)benzyl)amine (6d). Compound 4b (1180 mg, 5.43 mmol), compound 5b (504 mg, 2.78 mmol) and K2CO3 (2.00 g, 14.5 mmol) in acetonitrile (250 mL) yielded 6d as golden brown oily liquid (773 mg, 61.2%). Eluent for chromatography: hexane–chloroform 80:20 → 20:80 → 0:100. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.24 (d, J = 6.6 Hz, 18H), 3.27 (sep, J = 6.6 Hz, 3H), 3.81 (s, 6H), 7.12–7.15 (m, 3H), 7.19–7.21 (m, 3H), 7.36 (dd, J = 7.6 Hz, 1.2 Hz, 1H), 7.75 (dd, J = 7.5 Hz, 1.2 Hz, 1H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 23.23, 38.30, 56.07, 126.82, 126.93, 129.38, 131.98, 135.13, 141.13. HRMS: calculated for C30H39NS3 510.2278 [M + H]+; found 510.2327. Elemental analysis: calcd C 70.67, H 7.71, N 2.75, S 18.87; found C 70.40, H 8.03, N 2.56, S 19.07.
N-(2-(tert-Butylthio)benzyl)-N-(2-(methylthio)benzyl)-1-(2-(methylthio)phenyl)methanamine (6e). Compound 4a (451 mg, 2.08 mmol), compound 5c (206 mg, 1.06 mmol) and K2CO3 (800 mg, 5.79 mmol) in acetonitrile (100 mL) gave 6e as white solid (423 mg, 87.0%). Eluent for chromatography: hexane–chloroform 50:50 → 10:90. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.23 (s, 9H), 2.40 (s, 6H), 3.71 (s, 4H), 3.99 (s, 2H), 7.12–7.17 (m, 7H), 7.31–7.35 (m, 1H), 7.49 (dd, J = 7.8 Hz, 1.4 Hz, 1H), 7.67 (d, 7.2 Hz, 2H), 7.85 (dd, J = 7.8 Hz, 1.1 Hz, 1H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 16.11, 31.19, 47.32, 55.87, 56.82, 124.98, 125.53, 126.45, 127.27, 128.90, 129.11, 129.64, 132.32, 137.72, 137.74, 138.86, 144.89. HRMS: calculated for C27H33NS3 468.1809 [M + H]+; found 468.1855. Elemental analysis: calcd C 69.33, H 7.11, N 2.99, S 20.57; found C 68.65, H 7.26, N 2.89, S 20.59.
N,N-Bis(2-(tert-butylthio)benzyl)-1-(2-(methylthio)phenyl)-methanamine (6f). Compound 4c (1060 mg, 4.09 mmol), compound 5a (415 mg, 2.71 mmol) and K2CO3 (1.21 g, 8.76 mmol) in acetonitrile (150 mL) yielded 6f as golden brown oily liquid (542 mg, 51.9%). Eluent for chromatography: hexane–chloroform 50:50 → 0:100. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 1.22 (s, 18H), 2.40 (s, 3H), 3.68 (s, 2H), 3.95 (s, 4H), 7.11–7.16 (m, 5H), 7.32–7.35 (m, 2H), 7.49 (d, J = 7.8 Hz, 2H), 7.64 (d, J = 7.5 Hz, 1H), 7.84 (d, J = 7.7 Hz, 2H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 16.12, 31.21, 47.31, 55.92, 56.84, 124.96, 125.55, 126.43, 127.26, 128.92, 129.12, 129.66, 132.37, 137.86, 137.92, 138.89, 145.16. HRMS: calculated for C30H39NS3 510.2278 [M + H]+; found 510.233. Elemental analysis: calcd C 70.67, H 7.71, N 2.75, S 18.87; found C 70.09, H 8.01, N 2.65, S 19.12.
Tris(2-(tert-butylthio)benzyl)amine (6g). Compound 4c (541 mg, 1.83 mmol), compound 5c (261 mg, 1.33 mmol) and K2CO3 (810 mg, 5.86 mmol) in acetonitrile (100 mL) resulted in 6g as a white solid (386 mg, 76.5%). Eluent for chromatography: hexane–chloroform 50:50 → 10:90. 1H-NMR (CDCl3, 500 MHz) δ (ppm): 4.0 (s, 27H), 3.92(s, 6H), 7.13–7.16 (m, 3H), 7.33–7.35(s, 3H), 7.49 (dd, J = 7.7 Hz, 1.2 Hz, 3H), 7.82 (dd, J = 7.8 Hz, 1.2 Hz, 3H). 13C-NMR (CDCl3, 125 MHz) δ (ppm): 31.20, 42.27, 56.71, 126.38, 129.10, 129.59, 132.41, 138.89, 145.37. HRMS: calculated for C33H45NS3 552.2748 [M + H]+; found = 552.2795. Elemental analysis: calcd C 71.81, H 8.22, N 2.54, S 17.43; found C 71.49, H 8.49, N 2.48, S 17.70.
Extraction experiments
In a 50 mL centrifuge tube, an aliquot of chloroform containing 1 × 10−5 to 2 × 10−3 M of each ligand (6a–g) and an equal volume of aqueous buffer solution containing 10 mg L−1 of Ag+ as AgNO3, were mixed. Buffer solutions were prepared from 10 mM acetic acid and the pH values were adjusted by varying the ratio of potassium nitrate and potassium hydroxide, with the total potassium ion concentration adjusted to 100 mM. The centrifuge tubes were then shaken at 25 °C over 3 h. After the two phases were separated by centrifugation, the pH values (pH electrode) and the metal concentrations (ICP-AES) in the aqueous phase were measured. Moreover, the organic phase was shaken at 25 °C over 3 h with 1 M HNO3 as back extraction solvent. After separation in the same way, Ag+ concentrations in the aqueous phase (back extraction phase) were measured.
The extraction efficiencies, back extraction efficiencies, and total extraction efficiencies were determined based on the Ag+ concentrations in each set of aqueous phases according to the following equations,
| Extraction efficiency (%) = (Cw − Ce)/Cw × 100 |
| Back extraction efficiency (%) = C′e/(Cw − Ce) × 100 |
| Total extraction efficiency (%) = C′e/Cw × 100 |
where
Cw,
Ce, and
C′e correspond to the Ag
+ ion concentrations in the aqueous phase before extraction, after extraction, and in the back extraction aqueous phase, respectively.
Extraction efficiencies for other ions were evaluated according to the same procedure as described for Ag+.
Results and discussion
Molecular design and synthesis
As sterically demanding groups in the sidearms, the Ag+ extracting ligands include combinations of methyl and isopropyl, or methyl and tert-butyl substituents. The target compounds were obtained by the SN2 reactions of 2 equivalents of bromo derivatives and 1 equivalent of amine derivatives (Scheme 1). This synthesis method enables the access to ligands having combinations of various substituents. Though there are some literature reports about tripodal ligands with three identical coordinating arms for extraction, to the best of our knowledge, the effect of combinations of sidearms in tripodal ligands has not yet been evaluated in terms of extraction and back-extraction efficiencies. In this work, the effect of the combination of sterically demanding groups on the extraction and back-extraction efficiency was examined. All ligands have multiple sulfur atoms as “soft” bases and efficiently extract Ag+ ions as “soft” acid according to the HSAB concept.
 |
| | Scheme 1 (a) CH3I, i-PrBr or t-BuOH (b) BH3·THF, THF (c) PBr3, Et2O (d) NH3 aq., EtOH (e) K2CO3, CH3CN. | |
Determination of the extraction constant and confirmation of 1:1 complex stoichiometry
Extraction behavior of Ag+ with ligands in chloroform: the distribution ratio (D) of the silver ion is defined as| |
 | (1) |
where CAg, org and CAg, aq. are the total concentrations of Ag+ ions in the organic and aqueous phases, respectively.
The extraction equilibrium can be formulated as
| | |
Ag+(aq.) + NO3−(aq.) + nLorg ⇌ [LnAgNO3]org
| (2) |
The extraction constant, Kex, of Ag+ with the ligand is expressed as
| |
 | (3) |
As the total concentration of Ag+ ions in the organic phase corresponds to [LnAgNO3], the distribution ratio of Ag+ can be rewritten as
| |
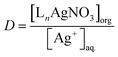 | (4) |
Substitution of eqn (3) into (4) results in the following
| | |
D/[NO3−]aq. = Kex[L]orgn
| (5) |
Taking the logarithm of eqn (5) yields
| | |
logD/[NO3−]aq. = logKex + nlog[L]org
| (6) |
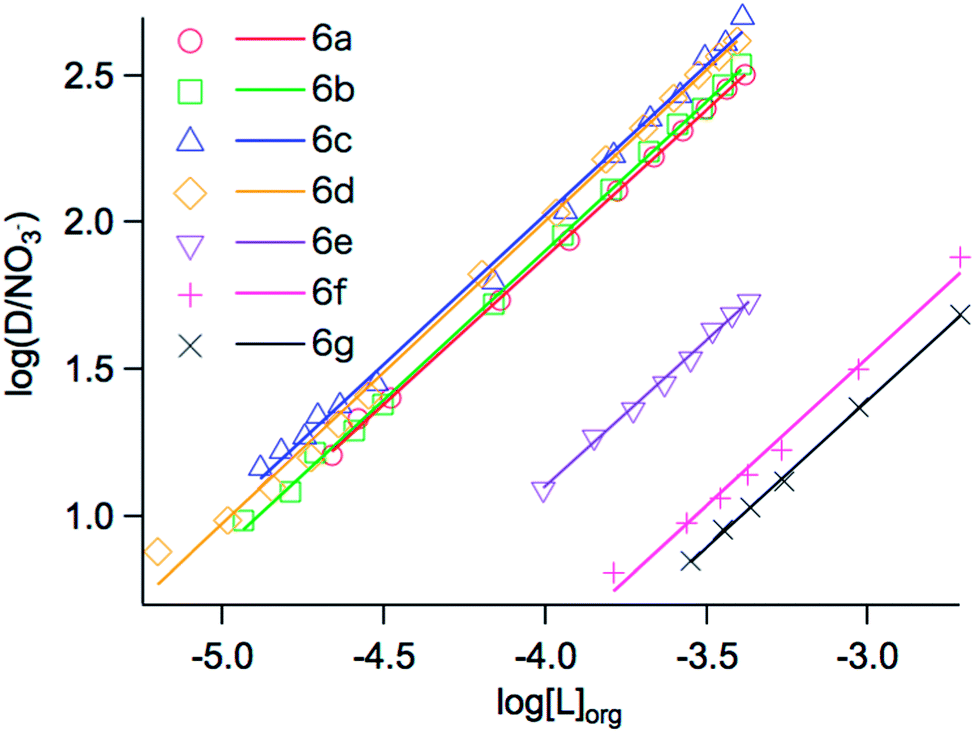
To confirm the complex stoichiometry with Ag+, as well as to estimate the extraction constant (Kex) of each ligand for Ag+, the distribution ratio D was determined as a function of the ligand concentration. Plots of log(D/[NO3−]aq.) versus the concentration of each ligand with different steric hindrance, log[L]org, at fixed pH of 4.0 are shown in Fig. 1. In all cases, straight lines with a slope of one are observed, confirming a 1:1 complex formation according to eqn (6). A schematic representation of a complex is shown in Scheme 2. This indicates that the stoichiometry of complexes formed with Ag+ is identical for all ligands, independent of the sidearms having different steric hindrance characteristics. ESI-MS mass spectra of mixtures of each ligand and AgClO4 also revealed the formation of a 1:1 complex through the metal coordination interaction, with two peaks (derived from Ag+ isotopes) assigned to the species [6a–g + Ag]+, respectively (shown in the ESI†). In addition, 1H and 13C-NMR spectra of 6a in the presence of AgNO3 showed changes of the chemical shifts compared to the spectra recorded in the absence of AgNO3 (shown in the ESI†). These results further demonstrate the interaction of the ligand with Ag+. Table 1 lists the extraction constants (Kex) of each ligand for Ag+, estimated according to eqn (6) from the intercept of the linear regression lines shown in Fig. 1. With an increasing number of t-Bu substituents in compounds 6a, 6e–g, Kex decreased from 5.89 to 5.10, 4.57 and 4.40, respectively. This might be the result of increasing binding distances between Ag+ and the donor S atoms, as well as the hindrance of binding between the counter anion and Ag+, caused by the bulky t-Bu substituent directly connected to the S atoms. On the other hand, with increasing number of i-Pr substituents in compounds 6b–d, Kex slightly increased to 5.97, 6.04 and 6.12, respectively. The inductive effect of alkyl groups increases in the order of methyl, i-Pr, and t-Bu. It has been reported that the inductive effect sometimes outweighs the steric effect on the complex formation constant of a ligand with a metal ion.30 In analogy to that work, it is assumed that the steric hindrance effects of the i-Pr substituents in compounds 6a–d are outweighed by the inductive effects.
 |
| | Fig. 1 Plots of log(D/NO3−) versus log[L]org for the extraction of 10 mg L−1 Ag+ as AgNO3 with 6a–g into chloroform; aqueous phase: pH 4.0. | |
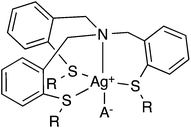 |
| | Scheme 2 General schematic representation of the structure of complexes formed between the tripodal ligands and Ag+. | |
Table 1 Extraction constants (Kex) of each ligand for Ag+, and slope and correlation coefficient (R2) of linear regression lines (data extracted from Fig. 1)
| |
6a |
6b |
6c |
6d |
6e |
6f |
6g |
| Slope |
1.00 |
1.02 |
1.01 |
1.03 |
1.00 |
1.01 |
1.00 |
| R2 |
0.999 |
0.999 |
0.996 |
0.996 |
0.996 |
0.987 |
0.999 |
| logKex |
5.89 |
5.97 |
6.04 |
6.12 |
5.10 |
4.57 |
4.40 |
Crystal structure studies
X-ray single crystal structural analysis was performed to determine the molecular structures of complexes formed between the ligands and Ag+. Since single crystals were more readily obtained as perchlorate salts than nitrate salts, AgClO4 was applied in crystallization experiments. Nevertheless, crystals sufficiently large for X-ray analysis were only achieved with ligands 6a and 6d. X-ray crystal structures of the two complexes Ag(6a)ClO4 and Ag(6d)ClO4 are shown in Fig. 2, together with selected structural parameters listed in Table 2. With respect to Ag(6d)ClO4, two crystallographically independent complexes (6dA and 6dB) were observed (Fig. 2b and c). The binding characteristics of ligands 6a and 6dA and 6dB with Ag+ are basically identical, with both compounds forming the same 5-fold coordinated complexes. Ag+ is coordinated by one N atom (Ag–N = 2.606 Å in 6a and 2.456 Å in 6d), three S atoms (Ag–S = 2.558 ± 0.006 Å in 6a and 2.54 Å in 6dA and 2.49 Å in 6dB) and one O atom from ClO4− (Ag–O = 2.576 Å in 6a and 3.29 Å in 6dA and 6dB). The distance between silver and each sulfur atom is slightly shorter than the mean (2.675 ± 0.015 Å) of 51 such bonds taken from the X-ray literature.31 Regardless of the substituent (Me or i-Pr), the Ag+–S bond lengths of 6a and 6d are nearly the same. This result implies that the sterically more demanding i-Pr moiety does not influence the binding ability of sulfur to Ag+, and it is adequate for the extraction constants (Kex values) of ligands 6a–d to slightly increase with increasing number of i-Pr groups according to the inductive effect. On the other hand, the distance between Ag+ and ClO4− of Ag(6a)ClO4 and Ag(6d)ClO4 are clearly different. This indicates that the presence of a sterically hindering substituent influences the distance between Ag+ and the counter anion.
 |
| | Fig. 2 X-ray single crystal structures of (a) 6a and (b) 6dA (c) 6dB complexing Ag+. | |
Table 2 Selected bond lengths for 6a and 6d complexing Ag+
| 6a |
6dA |
6dB |
| Ag(1)–S(3) |
2.5665(8) |
Ag(1)–S(1) |
2.54(1) |
Ag(1)–S(1) |
2.49(1) |
| Ag(1)–S(4) |
2.5593(8) |
Ag(1)–S(1) |
2.54(1) |
Ag(1)–S(1) |
2.487(9) |
| Ag(1)–S(5) |
2.548(1) |
Ag(1)–S(1) |
2.542(7) |
Ag(1)–S(1) |
2.49(2) |
| Ag(1)–N(7) |
2.606(2) |
Ag(1)–N(1) |
2.456(4) |
Ag(1)–N(1) |
2.456(4) |
| Ag(1)–O(38) |
2.576(2) |
Ag(1)–O(1) |
3.29(1) |
Ag(1)–O(1) |
3.29(1) |
Extraction and back-extraction behavior
The extraction efficiencies (%) of Ag+ with each ligand as a function of equilibrium pH in the aqueous phase are shown in Fig. 3. Ag+ was efficiently extracted with ligands 6a–d (increasing number of sterically demanding i-Pr groups) independent of the pH of the aqueous phase. The extraction efficiencies of Ag+ with 6e–g (increasing number of t-Bu groups) decreased with decreasing pH. This behavior was particularly pronounced for 6f and 6g. The results in Fig. 4 show the extraction efficiencies (%) of 6a for various other ions as a function of equilibrium pH in the aqueous phase. Independent of the pH of the aqueous phase, no significant extraction was observed for most ions, with the exception of Hg2+, which was extracted at an efficiency of about 8%. According to the HSAB concept, this extraction can be attributed to the relatively strong affinity of the sulfur atoms, characterized as ‘soft’ bases, for the ‘soft’ acid Hg2+. However, in accordance with the only limited extraction efficiency for Hg2+, these results demonstrate that ligand 6a has a suitably high selectivity for extraction of Ag+. The extraction efficiencies (%) of Ag+ with each ligand under identical pH condition of the aqueous phase (pH 4.0) are shown in Fig. 5a. The values of the extraction constants (Table 1) are directly reflected in the extraction efficiencies. In the case of ligands 6b–d (with i-Pr groups), the extraction efficiency was constantly high. In the case of ligands 6e–g (with t-Bu groups), the extraction efficiency decreased with increasing number of t-Bu groups. The back-extraction efficiencies (%) of Ag+ with each ligand for a 1 M HNO3 aqueous phase are shown in Fig. 5b. In the case of compound 6a, the back-extraction efficiency was only about 35% and not sufficient. This might be attributed to the strong interaction of S and Ag+.32 In the case of ligands 6b–d, the back-extraction efficiency increased with increasing number of i-Pr groups.
 |
| | Fig. 3 Extraction of Ag+ with various ligands as a function of the equilibrium pH of the aqueous phase; ligand concentration 1 mM. | |
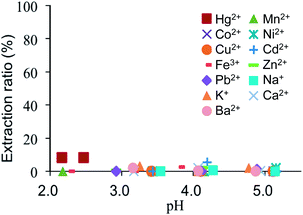 |
| | Fig. 4 Extraction of various ions with 6a as a function of the equilibrium pH of the aqueous phase; ligand concentration 1 mM. | |
 |
| | Fig. 5 (a) Extraction, (b) back-extraction, and (c) total-extraction efficiencies of Ag+ with various ligands; aqueous phase: pH 4.0; organic phase: ligand concentration 1 mM; back-extraction solvent: 1 M HNO3 aqueous solution. | |
Extraction and back-extraction efficiencies have been investigated using nitrate salts, due to their solubility in water over a wide concentration range, while X-ray analysis was done on complexes with perchlorate as the counter anion, because more ready crystallization. For comparison purposes, some extraction and back-extraction experiments were performed with AgClO4. The extraction efficiency and back-extraction efficiency (%) of Ag+ as AgNO3 or AgClO4 with 6d under identical pH condition of the aqueous phase (pH 4.0) are shown in Table 3. No significant differences in the extraction and back-extraction efficiencies of AgNO3 and AgClO4 were observed.
Table 3 Counter anion dependent extraction and back-extraction efficiencies of 6a and 6d for Ag+
| Ligand |
Anion |
Extraction |
Back-extraction |
Total-extraction |
| 6a |
NO3 |
98.8% |
35.4% |
35.0% |
| ClO4 |
99.0% |
35.7% |
35.3% |
| 6d |
NO3 |
99.4% |
74.5% |
74.0% |
| ClO4 |
98.4% |
72.1% |
71.0% |
From X-ray single crystal structural analysis of complexes formed with 6a and 6d, the distance between the Ag+ ion and the counter anion was larger in the complex of 6d than of 6a. Therefore, the difference of the distance between Ag+ and the counter anion might affect the back-extraction efficiencies. Similar to ligands 6b–d with increasing number of i-Pr groups, the back-extraction efficiency of compounds 6e–g increased with increasing number of t-Bu groups. These results again reflect the decreasing values of the extraction constants (Table 1), with increasing number of t-Bu groups.
The total extraction efficiencies (%) combining the extraction and back-extraction processes of Ag+ with each ligand are shown in Fig. 5c. With increasing number of i-Pr substituents (6a–d), the total extraction efficiencies increased up to 74%. The total extraction efficiencies decreased down to 57% with increasing number of t-Bu substituents. The total extraction efficiency of 6d was highest among the synthesized ligands (6a–g), because of the relatively high efficiency of both extraction and back-extraction. These results indicate that the steric hindrance of the alkyl-substituted thioether arms of the tripodal ligands affects the total extraction efficiency.
The reusability of ligands (6a and 6d) was evaluated as shown in Fig. 6. The total extraction efficiency of both ligands is hardly altered after three repeated cycles. These results indicate that these ligands are applicable to the repeated extraction of Ag+.
 |
| | Fig. 6 Total extraction efficiencies for the extraction of 200 mg L−1 Ag+ as AgNO3 with 6a or 6d; aqueous phase: pH 4.0; organic phase: ligand concentration 1 mM; back-extraction solvent: 1 M HNO3. | |
Conclusions
A series of tripodal ligands for Ag+ with thiophenylether sidearms carrying different sterically demanding groups have been successfully synthesized. The extraction and back-extraction efficiencies of these ligands depend on the number and size of sterically hindering groups. The extraction efficiencies decrease with increasing number of t-Bu groups. The back-extraction efficiencies increase with increasing number of i-Pr and t-Bu groups. The total extraction efficiency of ligand 6d having three i-Pr groups was highest in the new series of tripodal ligands (6a–g). These results indicate that the extraction efficiencies and the back-extraction efficiencies are controlled by the combination of sterically hindering groups present in the ligand binding arms. It is expected that these findings can be applied for the design of new metal extractants.
Notes and references
- Z. Dai and J. W. Canary, New J. Chem., 2007, 31, 1708–1718 RSC.
- M. Schatz, M. Becker, F. Thaler, F. Hampel, S. Schindler, R. R. Jacobson, Z. Tyeklar, N. N. Murthy, P. Ghosh, Q. Chen, J. Zubieta and K. D. Karlin, Inorg. Chem., 2001, 40, 2312–2322 CrossRef CAS PubMed.
- N. Wei, N. N. Murthy, Q. Chen, J. Zubieta and K. D. Karlin, Inorg. Chem., 1994, 33, 1953–1965 CrossRef CAS.
- J. T. Hoffman and C. J. Carrano, Inorg. Chim. Acta, 2006, 359, 1248–1254 CrossRef CAS PubMed.
- H. W. Yim, L. M. Tran, E. D. Dobbin, D. Rabinovich, L. M. Liable-Sands, C. D. Incarvito, K.-C. Lam and A. L. Rheingold, Inorg. Chem., 1999, 38, 2211–2215 CrossRef CAS PubMed.
- R. Wietzke, M. Mazzanti, J.-M. Latour, J. Pécaut, P.-Y. Cordier and C. Madic, Inorg. Chem., 1998, 37, 6690–6697 CrossRef CAS PubMed.
- N. Hirayama, Y. Horita, S. Oshima, K. Kubono, H. Kokusen and T. Honjo, Talanta, 2001, 53, 857–862 CrossRef CAS.
- R. J. Warr, A. N. Westra, K. J. Bell, J. Chartres, R. Ellis, C. Tong, T. G. Simmance, A. Gadzhieva, A. J. Blake, P. A. Tasker and M. Schroder, Chem. – Eur. J., 2009, 15, 4836–4850 CrossRef CAS PubMed.
- K. Matloka, A. K. Sah, M. W. Peters, P. Srinivasan, A. V. Gelis, M. Regalbuto and M. J. Scott, Inorg. Chem., 2007, 46, 10549–10563 CrossRef CAS PubMed.
- A. Pellissier, Y. Bretonniere, N. Chatterton, J. Pecaut, P. Delangle and M. Mazzanti, Inorg. Chem., 2007, 46, 3714–3725 CrossRef CAS PubMed.
- C. Kar, M. Deb Adhikari, A. Ramesh and G. Das, RSC Adv., 2012, 2, 9201–9206 RSC.
- A. Sengupta, P. K. Mohapatra, M. Iqbal, W. Verboom, J. Huskens and S. V. Godbole, RSC Adv., 2012, 2, 7492–7500 RSC.
- H. Okamura, A. Ikeda-Ohno, T. Saito, N. Aoyagi, H. Naganawa, N. Hirayama, S. Umetani, H. Imura and K. Shimojo, Anal. Chem., 2012, 84, 9332–9339 CAS.
- B. Swain, J. Jeong, S.-k. Kim and J.-c. Lee, Hydrometallurgy, 2010, 104, 1–7 CrossRef CAS PubMed.
- R. Banda, H. S. Jeon and M. S. Lee, Hydrometallurgy, 2012, 121–124, 74–80 CrossRef CAS PubMed.
- A. H. Bond, M. L. Dietz and R. Chiarizia, Ind. Eng. Chem. Res., 2000, 39, 3442–3464 CrossRef CAS.
- K. Ohto, Solvent Extr. Res. Dev., Jpn., 2010, 17, 1–18 CAS.
- M. Iwakuma, T. Ohshima and Y. Baba, Solvent Extr. Res. Dev., Jpn., 2008, 15, 21–35 CAS.
- H. Narita, K. Morisaku and M. Tanaka, Chem. Commun., 2008, 5921–5923 RSC.
- R. G. Pearson, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- I. Vujasinović, J. Veljković, K. Molcanov, B. Kojić-Prodić and K. Mlinarić-Majerski, J. Org. Chem., 2008, 73, 9221–9227 CrossRef PubMed.
- R. Alberto, W. Nef, A. Smith, T. A. Kaden, M. Neuburger, M. Zehnder, A. Frey, U. Abram and P. A. Schubiger, Inorg. Chem., 1996, 35, 3420–3427 CrossRef CAS PubMed.
- T. Nabeshima, K. Nishijima, N. Tsukada, H. Furusawa, T. Hosoya and Y. Yano, J. Chem. Soc., Chem. Commun., 1992, 1092–1094 RSC.
- P. A. Vigato, S. Tamburini and L. Bertolo, Coord. Chem. Rev., 2007, 251, 1311–1492 CrossRef CAS PubMed.
- A. T. Yordanov, B. R. Whittlesey and D. M. Roundhill, Inorg. Chem., 1998, 37, 3526–3531 CrossRef CAS PubMed.
- N. Singh and G. Hundal, J. Inclusion Phenom. Macrocyclic Chem., 2005, 52, 253–259 CrossRef CAS.
- L. G. A. van de Water, F. ten Hoonte, W. L. Driessen, J. Reedijk and D. C. Sherrington, Inorg. Chim. Acta, 2000, 303, 77–85 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- H. V. Huynh, C. H. Yeo and Y. X. Chew, Organometallics, 2010, 29, 1479–1486 CrossRef CAS.
- K. V. Damu, H. Maumela, R. D. Hancock, J. C. A. Boeyens and S. M. Dobson, J. Chem. Soc., Dalton Trans., 1991, 2717–2721 RSC.
- S. S. Lee, I. Yoon, K.-M. Park, J. H. Jung, L. F. Lindoy, A. Nezhadali and G. Rounaghi, J. Chem. Soc., Dalton Trans., 2002, 2180–2184 RSC.
- H. W. Mbatia, D. P. Kennedy and S. C. Burdette, Photochem. Photobiol., 2012, 88, 844–850 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR-spectra, HRMS spectra, ESI-MS spectra. CCDC 936706 and 936707. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c3ra45700a |
| ‡ Present address: Division of Physical Pharmaceutical Chemistry, Faculty of Pharmacy, Keio University, 1-5-30 Shibakoen, Minato-ku, Tokyo 105-8512, Japan. |
| § These authors have equally contributed to this work. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence