
Na2FeP2O7 as a positive electrode material for rechargeable aqueous sodium-ion batteries†
Abstract
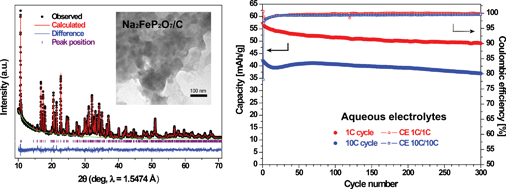
An iron-based pyrophosphate compound, Na2FeP2O7, is investigated as a positive electrode material for aqueous sodium-ion batteries for the first time. The high rate capability and good cyclability of this material in aqueous electrolytes are advantageous for low-cost and safe battery systems.
 Please wait while we load your content...
Please wait while we load your content...

