
Magnetic nanoparticles incorporated on functionalized mesoporous silica: an advanced electrochemical sensor for simultaneous determination of amiodarone and atenolol†
Abstract
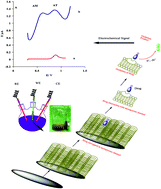
A new chemically modified carbon paste electrode is constructed based on magnetic (Fe2O3) mobile crystalline material-41 grafted with 3-aminopropyl groups (MCM-41–nPrNH2), a kind of mesoporous silica with large surface area (362 m2 g−1). For the first time, the electrode was evaluated as an electrochemical sensor for simultaneous determination of amiodarone and atenolol in aqueous solutions. The measurements were carried out by application of the differential pulse voltammetry method in Briton Robinson buffer solution with pH 4.00. Fe2O3 loaded in MCM-41–nPrNH2 can increase anodic peak currents by adsorption of amiodarone and atenolol on the electrode surface. The prepared electrode shows voltammetric responses with good selectivity for amiodarone and atenolol in optimal conditions, which makes it suitable for simultaneous determination of these drugs.
 Please wait while we load your content...
Please wait while we load your content...

