
Influence of different methylene units on the performance of rhodanine organic dyes for dye-sensitized solar cells†
Abstract
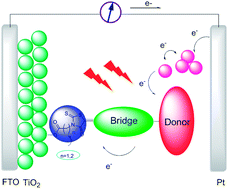
In D–π–A structured organic dyes, rhodanine acetic acid and its derivates have served as electron acceptors successfully. In this work, two single rhodanine organic dyes with different methylene units have been synthesized and applied in dye-sensitized solar cells (DSSCs). The photophysical, electrochemical and photovoltaic properties have been studied systematically. The results show that dye with double methylene units produces a higher photon-to-electron conversion efficiency (η) of 4.5% than that of a single methylene unit dye with a η of 4.1%. Also, it is interesting to notice that the lengthened distance between electron acceptor and TiO2 surface does not decrease the short-circuit current density (Jsc) and IPCE response for both structured dyes. On the contrary, rhodanine dye bearing double methylene units exhibits a better Jsc and IPCE response than the single methylene unit. The explanation for this trend is probably due to the fact that increased methylene units could suppress regeneration between the injected electron and the oxidized dye effectively.
 Please wait while we load your content...
Please wait while we load your content...

