Catalytic performance of highly dispersed Ni/TiO2 for dry and steam reforming of methane
Vijay M. Shinde and
Giridhar Madras*
Department of Chemical Engineering, Indian Institute of Science, Bangalore-560 012, India. E-mail: giridhar@chemeng.iisc.ernet.in; Fax: +91-80-23601310; Tel: +91-80-22932321
First published on 6th November 2013
Abstract
The present study reports a sonochemical-assisted synthesis of a highly active and coke resistant Ni/TiO2 catalyst for dry and steam reforming of methane. The catalyst was characterized using XRD, TEM, XPS, BET analyzer and TGA/DTA techniques. The TEM analysis showed that Ni nanoparticles were uniformly dispersed on TiO2 surface with a narrow size distribution. The catalyst prepared via this approach exhibited excellent activity and stability for both the reactions compared to the reference catalyst prepared from the conventional wet impregnation method. For dry reforming, 86% CH4 conversion and 84% CO2 conversion was obtained at 700 °C. Nearly 92% CH4 conversion and 77% CO selectivity was observed under a H2O/CH4 ratio of 1.2 at 700 °C for the steam reforming reaction. In particular, the present catalyst is extremely active and resistant to coke formation for steam reforming at low steam/carbon ratios. There is no significant modification of Ni particles size and no coke deposition, even after a long term reaction, demonstrating its potential applicability as an industrial reformate for hydrogen production. The detailed kinetic studies have been presented for steam reforming and the mechanism involving Langmuir–Hinshelwood kinetics with adsorptive dissociation of CH4 as a rate determining step has been used to correlate the experimental data.
1. Introduction
The steam reforming of methane is a well established industrial process for the production of hydrogen and synthetic gas.1,2 Ni-based catalysts are commonly used in industrial reforming reactions due to their low cost and reasonably good activity. However, these catalysts deactivate due to sintering and coke deposition.3 Therefore, excess steam (H2O/CH4 > 3) is introduced in the feedstock to prevent coke deposition. Though excess steam favors higher conversion, unnecessary generation of steam hampers the process economy and increases the H2/CO ratio. This makes the stream unsuitable for downstream processes such as Fischer-Tropsch synthesis.4 Furthermore, it increases the size of the reactor and the required amount of catalyst. It also leads to deactivation of the catalyst due to oxidation of Ni particles.5 Therefore, the inhibition of coke deposition under low H2O/CH4 ratio on the Ni-based catalysts is one of the biggest challenges in the steam reforming process.Several studies discuss enhancing the stability of Ni-based catalysts for the reforming reaction. Promoters such as alkaline earth oxide (MgO or CaO) are often used to lower the coking propensity and provide a higher stability against sintering.6,7 However, the addition of these promoters impedes the reduction of NiO leading to a decrease in the activity. It has been observed that promotion with K or Ca increases the formation of NiAl2O4 phase, which is reducible above 700 °C.8 Horiuchi et al. reported that the addition of alkaline metal suppressed the reforming activity of Ni with the markedly suppression of coke deposition for dry reforming reaction.7 Therefore, it is better to modify Ni-based catalysts without compromising their activity.
It is evident that the reducible supports (CeO2, TiO2) provide better stability and coke resistance in comparison with their non-reducible supports counterparts (Al2O3, SiO2).9–11 There is a direct correlation between oxygen storage capacity (OSC) and coke deposition propensity: the higher the OSC, the lower the coke deposition is on the catalyst.12 Ceria and modified ceria compounds are well known for their reversible exchange of lattice oxygen during the reaction.13–15 However, the CeO2 support is vulnerable to sintering and loses its OSC at high temperatures.16 TiO2 exhibits lower OSC compared to CeO2 but is stable at high temperatures. The dry reforming was studied over Ni supported on various supports, and specific activities followed the order: Ni/TiO2 > Ni/C > Ni/SiO2 > Ni/MgO.17 Therefore, TiO2 seems to be a good alternative support for CeO2 and Al2O3.
The activity of the catalyst often depends on the size and extent of metal dispersion.18,19 Small particles increases metal dispersion and also provides more steps/kinks on the surface.20,21 The energy barrier for methane dissociation, which is a rate determining step, over step sites is much lower than stair sites. Therefore, the rate of reaction increases with the extent of dispersion of the active phase.22,23 Further, small particles below a critical size have also been reported to be more resistant to coke formation and the spillover of steam on the support is a key parameter between particle size and rate of coke deposition.20,24 The highly dispersed metal particles also tend to minimize surface energy by increasing interaction with the support and hence minimizing sintering.22 Therefore, the synthesis of homogeneous and highly dispersed Ni nanosized particles with a significant metal support interaction is essential for stable performance.
The preparation method often influences the structure and morphology of the catalyst. Recently, it was shown that a sonochemical assisted method produces uniformly dispersed nanoparticles with a narrow size distribution in the range of 8–9 nm.25,26 Here, we report a sonochemical-assisted synthesis of a highly active and coke resistant Ni/TiO2 catalyst for dry and steam reforming of methane. In contrast to the conventional wet impregnation method, the metal precursor of the active phase and support are added together to an aqueous solution and irradiated using high intensity ultrasonic horn resulting in a very fine dispersion of the active phase. The catalytic performance of the material was investigated by performing dry and steam reforming reactions. High activity and stability was manifested for both the reactions. The intrinsic kinetics over a wide range of temperature was studied for the steam reforming reaction and the effect of inlet concentration of reactants and products on the rate of reaction was investigated. A Langmuir–Hinshelwood mechanism was used to correlate the experimental data.
2. Experimental
2.1 Synthesis and characterization
Titanyl nitrate (TiO(NO3)2) solution, and nickel nitrate (S.D Fine, India) were used as starting materials for the synthesis of 15% Ni/TiO2. A white colored TiO(OH)2 was precipitated by the controlled hydrolysis of 5 ml of titanium isopropoxide (Alfa Aesar, India) under ice cold (4 °C) conditions. 10 ml HNO3 was added to obtain a clear solution of TiO(NO3)2. In another precursor solution, 0.99 g of nickel nitrate was dissolved in 20 ml of distilled water and both the solutions were mixed together. The resulting solution was sonicated for 3 h using a high intensity Ti-horn probe of 25 mm diameter (50 KHz, and 125 W cm−2 at 60% efficiency). The powder was separated, washed with a water–ethanol mixture, and dried in a hot air oven at 120 °C. The catalytic activity of 15% Ni/TiO2 catalyst, synthesized by a conventional wet impregnation method, was compared with the catalyst synthesized via the sonication method. For this purpose, a pure known weight of TiO2 (prepared by sonication method) was dispersed in water and nickel nitrate equivalent to 15 wt% was added. Ni2+ ions were reduced to Ni metals by hydrazine hydrate (S.D Fine, India) solution at room temperature. The solid was separated and dried at 120 °C. Both the powders were further calcined at 700 °C for 1 h. The catalyst synthesized via the sonochemical method is designated as 15% Ni/TiO2 (sonic) while the catalyst synthesized via the impregnation method is designated as 15% Ni/TiO2 (imp).X-ray diffraction (XRD) patterns were recorded on a Philips X'Pert diffractometer with Cu-Kα radiation (λ = 1.54178 Å) in the 2θ range of 20–80°. Transmission electron microscopy (TEM; FEI Technai 20) was used to study the morphology and microstructures of the catalyst. The TEM specimen was prepared by dropping a trace amount of the sample dispersed in ethanol on a carbon coated grid (300 mesh). XPS spectra were recorded on a Thermo Scientific Multilab 2000 instrument with monochromatized Al-Kα X-rays (1486.6 eV). The binding energies were charge corrected using the C 1s peak observed at 285 eV. The BET area measurement was carried out with a Quantachrome NOVA 1000 gas adsorption analyzer. Prior to the measurement, the sample was degassed at 150 °C for 4 h under vacuum. The amount of carbon deposited during the long term stability test was determined using thermo-gravimetric analysis and differential thermal analysis (TGA/DTA). The experiment was performed on a Mettler Toledo thermal analyzer under O2 flow of 30 ml min−1 with a heating rate of 10 °C min−1.
2.2 Catalytic studies
Dry reforming of CH4 was studied in a fixed bed reactor under atmospheric pressure. The quartz reactor (4 mm ID and length of 30 cm) was heated in an electric furnace and the temperature of the bed was controlled by a K-type thermocouple positioned in the center of the catalyst bed. 75 mg of catalyst was packed between two glass wool plugs in the center of the reactor, the feed mixture consisting of 2% CH4, 2% CO2 and balance of N2 keeping the total flow rate at 100 ml min−1 (GHSV of 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 h−1 is based on the catalyst bed volume of 0.0628 cm3). The oxidized catalyst in air showed the formation of an inactive NiTiO3 phase and no activity for both the reactions. Therefore, the catalyst was reduced at 650 °C for 2 h with pure H2 at a flow of 20 ml min−1 before the reaction. The product was analyzed using an on-line gas chromatograph (Mayura Analytical Bangalore, India) equipped with a TCD and FID (incorporating a methanator). The conversions (X) and the H2/CO ratio were calculated as follows
500 h−1 is based on the catalyst bed volume of 0.0628 cm3). The oxidized catalyst in air showed the formation of an inactive NiTiO3 phase and no activity for both the reactions. Therefore, the catalyst was reduced at 650 °C for 2 h with pure H2 at a flow of 20 ml min−1 before the reaction. The product was analyzed using an on-line gas chromatograph (Mayura Analytical Bangalore, India) equipped with a TCD and FID (incorporating a methanator). The conversions (X) and the H2/CO ratio were calculated as follows
 | (1) |
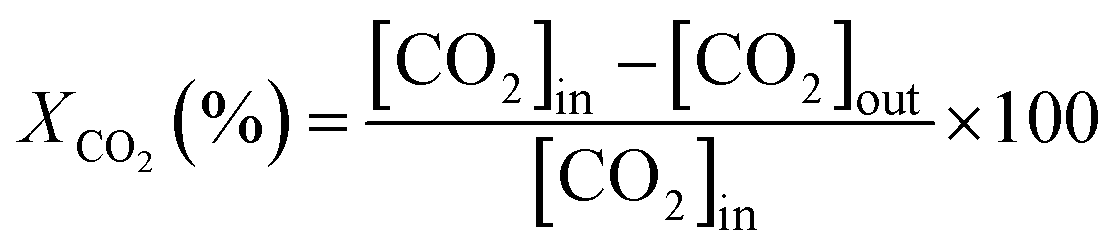 | (2) |
 | (3) |
The bracketed quantity represents the concentration of the component in the product stream. The activity of the catalyst was measured under a steady state at various temperatures. In order to ensure the steady state, the temperature of the reactor was set at the desired value and the gases were allowed to flow over the catalyst continuously. After 15 min, four readings at the same temperature were averaged. The temperature of the reactor was then set to the next high temperature and the same procedure was repeated. The average of the four readings was taken for the calculation and the standard deviation of the reported conversion was less than 3%. The experimental data was collected under the absence of any external and internal diffusion limitation.
The steam reforming reaction was carried over 150 mg of catalyst diluted by the required amount of glass beads. The feed mixture consisting of 3 vol% of CH4 and the balance of N2 was passed at rate of 100 ml min−1. This corresponds to a gas hourly space velocity of 48000 h−1 (based on the catalyst bed volume of 0.125 cm3). Water was fed to the steam generator using a HPLC pump (Waters 515) at a flow rate of 0.1 ml min−1 and the generated vapor (3.6 ml min−1) was mixed with the reaction mixture before entering the reactor. A moisture trap was kept at the outlet of the reactor to condense any water from the product gas stream. Prior to reaction, the catalyst was reduced in pure H2 with a flow rate of 20 ml min−1 for 2 h at 650 °C. The CH4 conversion (X) and CO selectivity were calculated as follows
 | (4) |
 | (5) |
The rate of formation of (CO + CO2) was nearly same to the rate of disappearance of CH4, which indicates that the rate of carbon formation was negligible over the catalyst.
3. Results and discussion
3.1 Structural studies
XRD patterns of both the catalysts before and after the steam reforming reaction are shown in Fig. 1. The 15% Ni/TiO2 (sonic) catalyst shows well resolved peaks characteristic of a rutile structure of TiO2. The peaks observed at 2θ = 76.4, 51.8 and 44.5° can be assigned to the (220), (200), (111) planes of Ni metal, respectively. However, 15% Ni/TiO2 (imp) catalyst shows peaks due to metallic Ni (111) along with anatase phases of TiO2. The small peak at 27.4° indicates that the small amount of the rutile phase is also present in 15% Ni/TiO2 (imp) catalyst. The average crystallite size of Ni was determined by the peak broadening of the (111) reflection in the XRD patterns, using the Scherrer formula, and was found to be 13 nm and 19 nm respectively, for the 15% Ni/TiO2 (sonic) and 15% Ni/TiO2 (imp) catalysts. Furthermore, the close observation of the XRD patterns of the 15% Ni/TiO2 (sonic) catalyst shows that the peak of the rutile TiO2 phase observed at 27.6° is shifted to higher values by 0.4° due to decrease in d-spacing. This shows partial substitution of Ni into TiO2 lattice during synthesis. In contrast, no shift in the XRD pattern of the 15% Ni/TiO2 (imp) catalyst was observed. Therefore, partial incorporation of Ni into TiO2 lattice is possible during synthesis.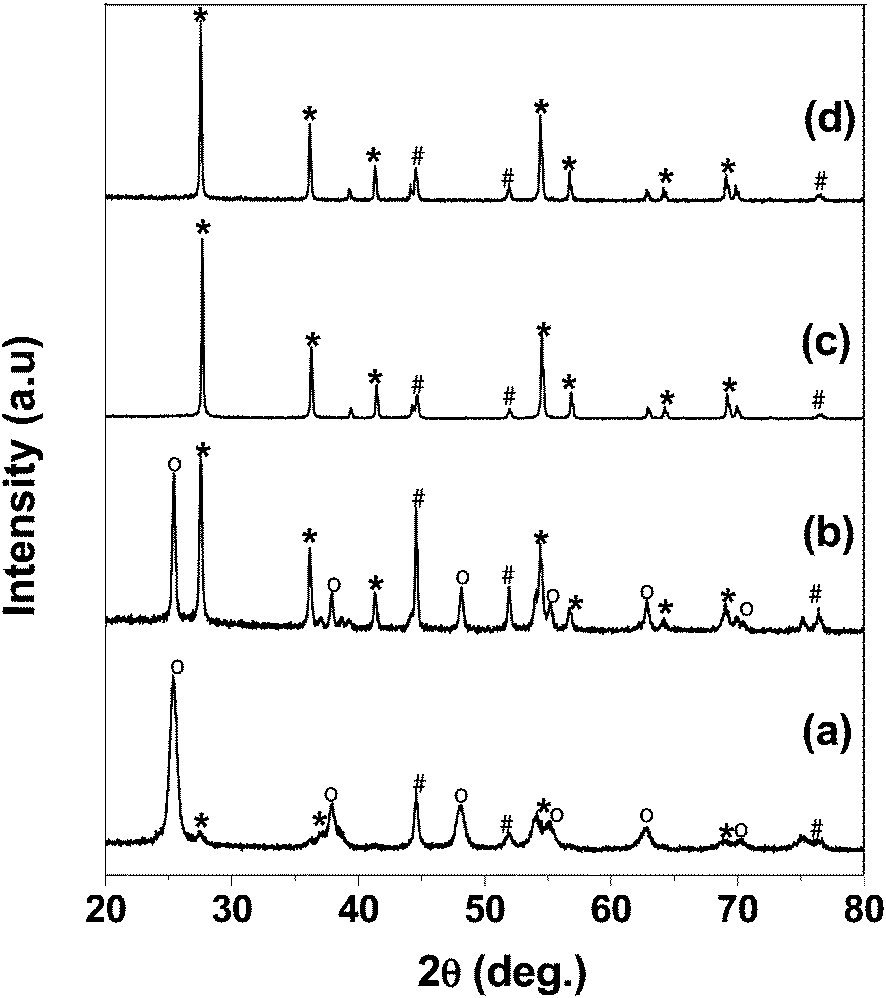 | ||
| Fig. 1 XRD patterns of (a) 15% Ni/TiO2 (imp) before reaction (b) 15% Ni/TiO2 (imp) after reaction, (c) 15% Ni/TiO2 (sonic) before reaction and (d) 15% Ni/TiO2 (sonic) after reaction. o anatase, * rutile, # Ni metal. | ||
It is well known that Ni/TiO2 catalysts deactivate due to the formation of the NiTiO3 phase and oxidation of the Ni species, which make the catalyst more difficult to reduce during the reaction. Therefore, the XRD patterns were also recorded after the reaction to observe the changes in the catalyst structure. The XRD patterns of the 15% Ni/TiO2 (sonic) shows that the rutile structure of TiO2 was retained after the reaction and reflections either due to NiO or NiTiO3 were not observed, indicating that the catalyst is stable and not oxidized during the reaction. For the Ni/TiO2 (imp) catalyst, peaks either due to NiO or NiTiO3 are also absent. However, the formation of the rutile phase was found to be high after the reaction.
Bright field images of the 15% Ni/TiO2 (sonic) and the 15% Ni/TiO2 (imp) catalyst are shown in Fig. 2. In the as synthesized 15% Ni/TiO2 (sonic) catalyst (Fig. 2(a)), Ni particles are spherical and uniformly distributed over the TiO2 support and no aggregation of particles was observed. The average particle size of the Ni species was 8–10 nm. After calcination at 700 °C, the size of the Ni nanoparticles slightly increased. The average particle size of Ni is about 14–16 nm, which is similar to the size calculated from the broadening of the XRD diffraction patterns. After 16 h of the steady state test reaction, the presence of appreciable carbon was not observed (Fig. 2(c)), which is an agreement with the TGA/DTA (∼0.6 wt%) results. The TEM image shows that the Ni particles retained physical contact with the TiO2 support and no considerable agglomeration of the Ni species was observed. However, the average particle size of the Ni metal is between 16 and 18 nm. This shows that there is no appreciable increase in the size of Ni particles (compared to calcinated catalyst) during the reaction. In contrast, the 15% Ni/TiO2 (imp) catalyst forms large nanoparticles compared to the 15% Ni/TiO2 (sonic) catalyst (Fig. 2(d)). The BET surface area for the catalyst before and after the steam reforming reaction was found to be 62 and 49 m2 g−1, respectively. This reduction in surface area may be due to an increase in the particles’ sizes.
 | ||
| Fig. 2 Bright field images of the 15% Ni/TiO2 (sonic) catalyst (a) as-synthesized (b) calcinated at 700 °C for 1 h (c) after reaction and (d) the 15% Ni/TiO2 (imp) catalyst. | ||
Fig. 3 shows XPS of both the catalysts. The NiO species is characterized by the peak at a binding energy of 854.6 eV along with the broad satellite at around 860 eV.27 A peak at a binding energy of 852.6 eV corresponds to Ni0 metal.28 The main Ni (2p) peak was deconvoluted corresponding to the Ni2+ and Ni0 states. Ti (2p3/2) binding energies are observed at ∼458.9 eV in both the catalysts corresponds to a Ti ion in the +4 state.29 The spectra of the spent catalyst were very broad, indicating the partial reduction of Ti4+ ions to the Ti3+ state. The Ti3+ ion in Ti2O3 is observed at 458.2 eV.29 It was found that the binding energy of the Ni (2p) and Ti (2p) peaks in the 15% Ni/TiO2 (sonic) catalyst were shifted slightly to higher values compared to the 15% Ni/TiO2 (imp) catalyst. The shift in binding energy of Ni (2p) in the 15% Ni/TiO2 (sonic) catalyst is due to the different chemical environment of the substituted Ni ions in the catalyst compared to that of the pure Ni ions in NiO. The metal-support interaction has previously been observed after steam treatment of a Ni/Al2O3 catalyst and a shift in binding energy of Ni 2p and Al 2p peaks towards higher values was observed after steam pretreatment.30 The surface concentration of Ni in both the catalysts was estimated from the intensities of the Ni (2p) and Ti (2p) peaks. The relative surface concentration was calculated from
 | (6) |
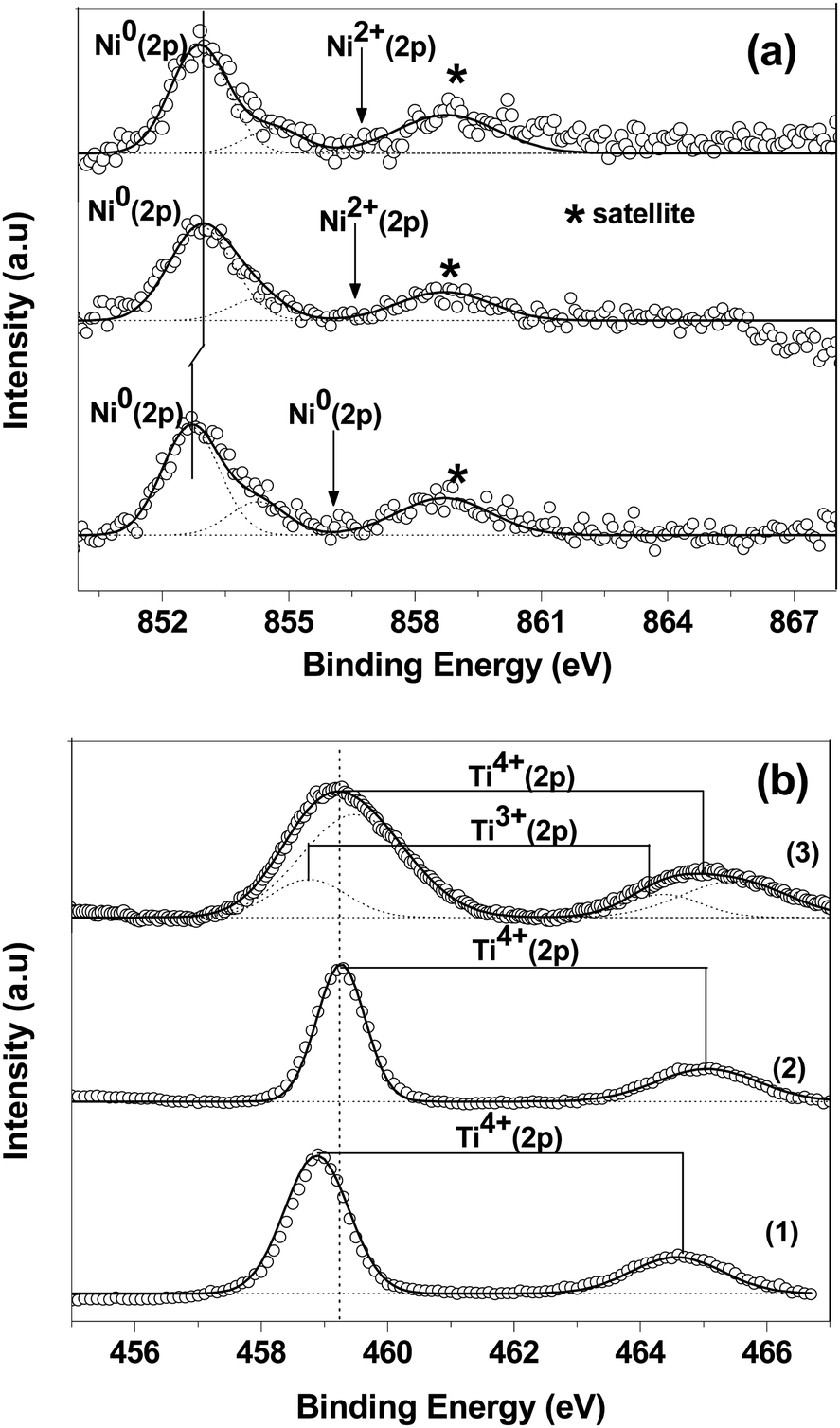 | ||
| Fig. 3 Core level XPS of (a) Ni 2p and (b) Ti 2p. The numbers (1), (2) and (3) represent spectra for the 15% Ni/TiO2 (imp) catalyst, and the 15% Ni/TiO2 (sonic) catalyst before and after the reaction, respectively. | ||
IM, λM, σM and DM are the integral intensity of the Ni (2p) and Ti (2p) peaks, mean escape depths of the respective photoelectrons, photoionization cross section, and geometric factor, respectively. The photoionization cross-section values and mean escape depths were taken from the literature.31,32 The geometric factor was taken as 1, since the maximum intensity in this spectrometer is obtained at 90°. The relative surface concentrations of Ni species obtained for 15% Ni/TiO2 (sonic) and 15% Ni/TiO2 (ionic) were 26% and 19% at, respectively, which is much higher than the 15 wt% (15 wt% corresponds to 17% at) taken in the preparation. After the reaction, the relative surface concentration of Ni species for 15% Ni/TiO2 (sonic) catalyst was found to be 18%.
3.2 Catalytic performance for dry reforming
The activity of the catalyst for the dry reforming was expressed in terms of conversion of the reactants. The variation of CH4 and CO2 conversions over the 15% Ni/TiO2 (sonic) catalyst with temperature is shown in Fig. 4(a). Both the conversions increased with increasing temperature. In addition, the CO2 conversion was similar to CH4 conversion, which indicates that the contribution of the reverse water gas shift reaction (CO2 + H2 ↔ CO + H2O) is negligible. Nearly 86% CH4 conversion and 84% CO2 conversion was observed at 700 °C. The catalysts with the low Ni loading, namely 5% and 10% over TiO2, were also synthesized and tested for the dry reforming reaction. Both CH4 and CO2 conversions were found to be lower than for the 15% Ni/TiO2 catalyst. At 700 °C, 31% CH4 and 34% CO2 conversion was obtained over the 5% Ni/TiO2 catalyst while only 59% CH4 and 56% CO2 conversion was obtained over the 10% Ni/TiO2 catalyst.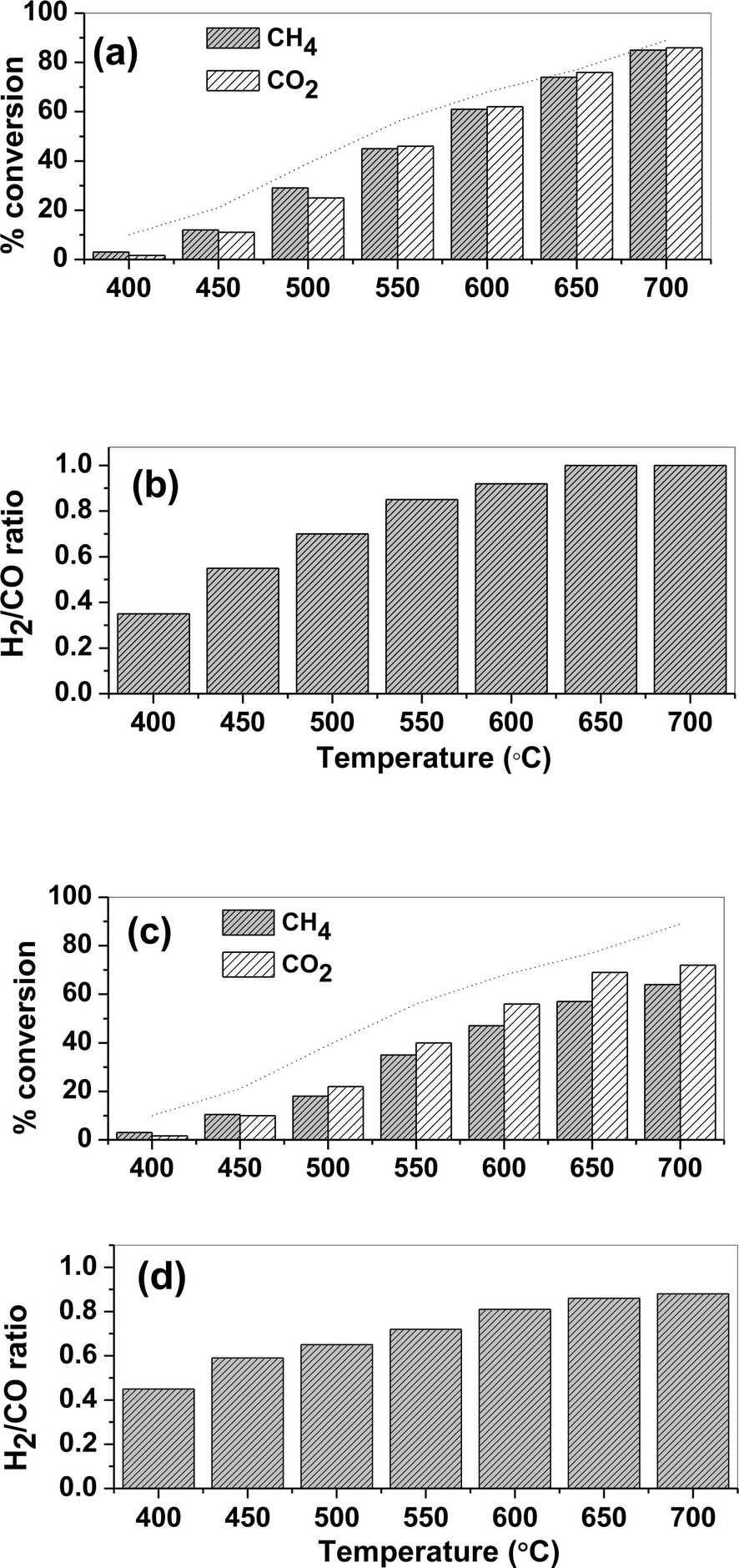 | ||
| Fig. 4 Catalytic activity for the dry reforming (a) CH4 conversion and CO2 conversion (b) H2/CO ratio as a function of temperature for 15% Ni/TiO2 (sonic) catalyst and (c) CH4 conversion and CO2 conversion (d) H2/CO ratio as a function of temperature for 15% Ni/TiO2 (imp) catalyst. | ||
The catalyst performance of 15% Ni/TiO2 (sonic) was also compared against the catalyst synthesized via the conventional wet impregnation method. Ni loading and amount of catalyst in both cases were kept constant. CH4 and CO2 conversions as a function of temperature over 15% Ni/TiO2 (imp) are depicted in Fig. 4(c). Nearly, 64% CH4 conversion and 72% CO2 conversion was obtained at 700 °C in the presence of the impregnated catalyst. At high temperature, the rate of reaction is controlled by diffusion of reactant. Therefore, the rate of reaction was expressed in mol g−1 s−1 and the performance of both the catalysts was compared at low temperatures. It was found that the reaction rates were higher for the 15% Ni/TiO2 (sonic) catalyst. Therefore, the catalyst synthesized by the sonication method exhibits higher activity than the catalyst synthesized via a conventional wet impregnation. The enhancement in reforming activities of the 15% Ni/TiO2 (sonic) catalyst is due to intimate contact between Ni and TiO2 support, as evidenced from XPS studies. It must be noted that XRD and TEM studies showed that Ni in the catalyst synthesized by the sonication method had smaller crystallites’ size and high metal dispersion. Therefore, the enhancement in the activity of the catalyst is related to the intimate contact of Ni and TiO2 support and the fine dispersion of the active species. Furthermore, despite an equimolar amount of CH4 and CO2 in the feed, CO2 conversion was higher compared to conversion of CH4 for the temperature range over 15% Ni/TiO2(imp) catalyst. This indicates that the extent of occurrence of the reverse water gas shift reaction is higher over this catalyst.
The H2/CO ratio is important for downstream processes. Fig. 4(b) and (d) show the H2/CO molar ratio, respectively, for the 15% Ni/TiO2 (sonic) and 15% Ni/TiO2 (imp) catalyst at various reaction temperatures. The H2/CO ratio increased with an increase in temperature for both the catalysts. In particular, the 15% Ni/TiO2 (sonic) catalyst reached a H2/CO ratio close to 1 above 650 °C whereas 15% Ni/TiO2 (imp) catalyst showed H2/CO ratio lower than 1. The low value of the H2/CO ratio (<1) is due to the strong contribution of the reverse water gas shift. The simultaneous occurrence of a reverse water gas shift reaction consumes additional H2 and produces extra CO, which lowers the H2/CO ratio.
3.3 Kinetic study of steam reforming
Fig. 5(a) shows the variation of CH4 conversion and CO selectivity with temperature over the 15% Ni/TiO2 (sonic) catalyst. Both CH4 conversion and CO selectivity increased with increases in temperature. A nearly 92% CH4 conversion with 77% CO selectivity was observed at 700 °C. The CH4 conversion below 400 °C was small and a sharp rise in the CH4 conversion was observed above 400 °C indicating a high energy barrier for this reaction. Furthermore, at low temperatures, CO selectivity is low, which implies a large contribution from the WGS (water gas shift) reaction. The effect of steam concentration on the CO selectivity is also depicted in Fig. 5(b). The CO selectivity decreased with an increase in the inlet steam concentration due to promotion of the WGS activity. | ||
| Fig. 5 Variation of (a) CH4 conversion and (b) CO selectivity with H2O/CH4 ratio at various temperatures over the 15% Ni/TiO2 (sonic) catalyst for the steam reforming reaction. | ||
The rate of reaction and activation energy for reforming over the 15% Ni/TiO2 (sonic) catalyst was measured by performing experiments with different amounts of catalyst loading keeping the total flow rate constant at 100 ml min−1. The mixture consisting of 3% of CH4, 3% of H2O and a balance of N2 with the total flow rate of 100 ml min−1 was used. All experiments were performed under isothermal and differential conditions at atmospheric pressure over a temperature range of 450–550 °C, and the rate of reaction at various temperatures was calculated using following equation:
 | (7) |
F, W and x are the flow of the gas in mol s−1, weight of the catalyst in g and fractional CH4 conversion, respectively. Fig. 6(a) shows the variation of W/FCH4 with the fractional conversion of CH4 at various temperatures. The plot of fractional conversion (x) with W/FCH4 is linear up to 40% conversion, and non linearity at higher CH4 conversion indicates that the differential reactor approach is not valid at high temperatures. Therefore, the rates of reaction were calculated from the slope of the linear portion. The variation of rate of reaction with temperature is shown in Fig. 6(b). The apparent activation energy was calculated from the Arrhenius plot (see inset of Fig. 6(b)) and found to be 105 kJ mol−1.
 | ||
| Fig. 6 (a) Variation of fractional conversion of CH4 with W/FCH4 and (b) rate of reaction as a function of temperature for steam reforming reaction over the 15% Ni/TiO2 (sonic) catalyst. | ||
The effect of concentration of CH4, steam and CO on the rate of reaction was also investigated to understand the reaction kinetics. The rate of reaction was measured in a differential reactor in the absence of any transport artifacts under atmospheric pressure. All of the experiments were carried out over 50 mg of catalyst with a total flow of 100 ml min−1 and the reactor temperature was varied such that the differential reactor approach (CH4 conversion ∼25%) was always maintained. The concentration of CH4 was varied between 1 and 4% keeping the steam concentration constant at 3.6%. For another set of experiments, the concentration of steam was varied between 1 and 8% keeping the methane concentration constant at 3%. The effect of CO concentration on the rate of the reaction was also independently examined. The concentration of CO was varied in the range of 0.25 to 0.75%, while the inlet concentrations of CH4 (3%) and steam (3.6%) were kept constant.
Fig. 7(a) shows the variation of rate of reaction with concentration of CH4 at various reaction temperatures. The rate of reaction increases with the concentration of CH4 at all temperatures. The order of reaction with respect to concentration of CH4 was determined by plotting the variation of rate of reaction with concentration on a log–log scale. In general, the reaction has a first order relationship with CH4 concentration for all temperatures, and it is consistent with rate determining step, namely CH4 chemisorption. Fig. 7(b) shows the variation of the rate of reaction with steam concentration. A strong negative effect of steam concentration on the rate of reaction was observed. This effect is attributed to an optimum concentration of steam and CH4 coverage on the catalyst surface. The negative order of reaction with steam concentration indicates that CH4 and steam undergo competitive adsorption on the catalyst for the same active sites. However, no maximum in the rate of reaction was observed when CH4 concentration was varied. This observation is in line with a reaction scheme in which the rate limiting step is the activation of CH4 molecules. The effect of inlet concentration of CO on the steam reforming is also depicted in Fig. 7(c). The rate of reaction decreases with increasing inlet concentration of CO. This effect is due to the chemisorption of CO interfering with CH4 chemisorption on the same active surface sites. The qualitative trends presented above elucidate the major mechanistic aspects of the steam reforming reaction. Further, the measured rates reflect intrinsic kinetics without any mass transfer effects. Therefore, these observations were used to propose the reaction mechanism for the steam reforming reaction.
 | ||
| Fig. 7 (a) The effect of concentration of CH4 on the rate of methane conversion at a constant steam concentration of 3.6%, (b) effect of H2O concentration on the rate of methane conversion at a constant methane concentration of 3% (c) effect of CO concentration on the rate of methane conversion at constant methane (3%) and steam concentration (3.6%), respectively. | ||
3.4 Kinetic model
The kinetics of steam reforming has been studied extensively over noble metals (Ru, Rh, Pt) and nickel-based catalysts. However, there is no agreement in the reaction mechanism and the corresponding rate expression. Several expressions including Langmuir–Hinshelwood, power laws, and expressions based on micro kinetic analysis have been proposed to describe the kinetics of reforming reactions.6,33 This is likely due to the different nature of the active species, identity of support, catalyst morphology (shape and size), different experimental conditions (synthesis method and testing condition etc.) and metal loading.20 In most of the studies, the dissociative methane adsorption reaction is assumed to be a rate determining step.8,32 Steam and dry reforming over Ru catalysts supported on various supports have been studied and a first order kinetic equation of CH4 concentration was proposed. No dependence on the nature of support, metal loading and concentration of H2O or CO2 was observed.19,34 The effect of various forms of support on the activity of the reaction has also been studied. Berman et al. have reported a negative reaction order in steam concentration, and speculated that the adsorption of steam occurs on Al2O3/MnOx support.35 A kinetic model involving CO2 interaction with the support was proposed for dry reforming of CH4 over a Ru/La2O3 catalyst.36 On the other hand, no influence of the support on the reaction rate has been reported.22,37The reforming reaction was studied over nickel foil at 900 °C at atmospheric pressure and the rate of reaction is satisfactory described by a simple first order equation (rate = kPCH4), where PCH4 is the partial pressure of the CH4 and k is the rate constant.38 However, a simple first order equation cannot be used to describe the kinetics of this reaction because the inlet partial pressure of H2O has a significant influence on the rate of reaction. A high CO coverage was observed over the catalyst during the reaction.5 The present study also showed the negative effect of the concentration of H2O and CO on the reaction. Therefore, the development of an improved model with the effect of concentration of steam and products on the overall rate of reaction is necessary. It should be noted that the kinetic models for methane reforming are quite different from each other and depend on the experimental conditions.20
Based on our observations and the available literature, we have proposed a set of elementary steps and the kinetic expression was derived using a Langmuir–Hinshelwood kinetic by considering the CH4 dissociative adsorption as the rate limiting step.39
 | (8) |
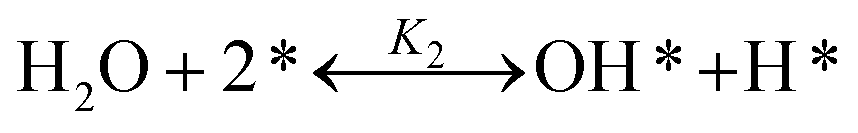 | (9) |
 | (10) |
 | (11) |
 | (12) |
 | (13) |
Eqn (8) represents the decomposition of CH4 into the adsorbed surface containing species (CH3*, CH2*, CH* and H*). The rate of hydrogen removal in subsequent steps is much higher than that of the initial C–H bond rupture. Therefore, subsequent H-abstraction from CH4 molecules is represented by a single equation. The negative order with respect to H2O shows that there is competitive adsorption between CH4 and steam for the same active sites. Therefore, eqn (8) and (9) assume that the activation of CH4 and steam compete for same active sites. The rapid steam adsorption gives the surface oxygen species (eqn (10)) and the carbon species formed on the surface. This reacts rapidly with the surface oxygen species resulting in product, CO (eqn (11)).40 Furthermore, a high WGS indicates that CO adsorption over the catalyst surface is considerable. The rate of reaction decreases with increasing inlet concentration of CO due to interference of CO with the CH4 chemisorption for the same surface sites (eqn (12)).
The rate of reaction is first order with respect to CH4 concentration and this implies that the dissociative adsorption of CH4 is a rate determining step. Therefore, while deriving the kinetic expression, it was explicitly assumed that the dissociative adsorption of CH4 (eqn (8)) is a rate limiting step. Solving the above set of elementary steps, the following rate expression is obtained.
 | (14) |
The above kinetic expression is consistent with the observations that the dissociative CH4 adsorption step, a rate determining step, and activation of H2O molecules takes place over a single metal surface atom without involvement of the support. The values of K1, K2 and K5 were obtained simultaneously using non-linear regression. Fig. 8 shows the predicted rate of reaction corresponding to the experimental observed reaction rate. The model without an inhibition term due to CO adsorption was also tried to fit the data but this resulted in poor fitting. Therefore, the adsorption of CO over the catalyst is substantial and cannot be ignored. The high activity of the catalyst for WGS reaction also demonstrates this fact. The optimized values for K1, K2 and K5 are 68exp(−4400/RT), 5400exp(4900/RT) and 1200exp(−1600/RT), respectively.
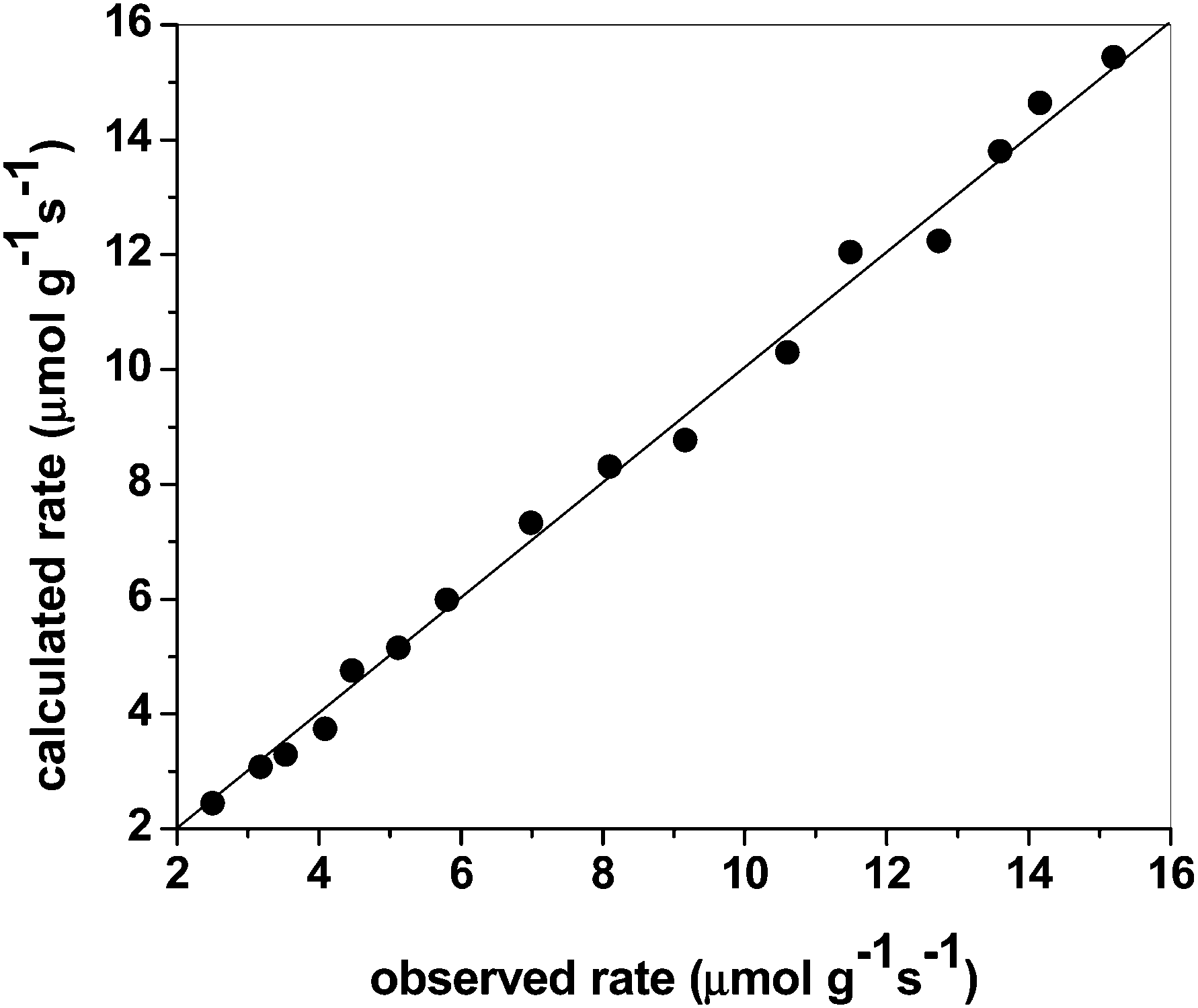 | ||
| Fig. 8 Comparison between experimentally measured rate and calculated rate from the model for steam reforming reaction. | ||
4. Stability and deactivation studies
The dependence of the catalytic activity on time on stream over the 15% Ni/TiO2 (sonic) catalyst was studied at 650 °C for 16 h for both dry and steam reforming reactions. Fig. 9(a) shows the variation of CH4, CO2 conversions and H2/CO ratio over 16 h for the dry reforming reaction. The stable conversion values for CH4 and CO2 were 65% and 67%, respectively. A small drop (about 2–3%) in both the conversions was observed for the initial few hours, which might be due to some modification of the catalyst surface causing instability in the carbon deposition.41 However, the H2/CO ratio (∼0.96) was nearly stable over the 16 h reaction period. The stability of the 15% Ni/TiO2 (imp) catalyst was also studied at 650 °C for 16 h for the dry reforming reaction. Fig. 9(b) shows the variation of CH4, CO2 conversions and H2/CO ratio over 16 h. The time on stream test shows that 15% Ni/TiO2 (imp) catalyst is less stable and more than 30% loss of initial activity was observed during first 4 h of reaction. Fig. 9(c) shows the stability for steam reforming reaction. There was a slight decrease in CH4 conversion and CO selectivity for the initial period. However, a stable CH4 conversion (∼75) and CO selectivity (∼62) was observed after 4 h. Therefore, the catalyst has high activity and excellent stability for both the reactions. | ||
| Fig. 9 Time on stream (a) CH4, CO2 conversion and H2/CO ratio for dry reforming of methane over the 15% Ni/TiO2 (sonic) catalyst, (b) CH4, CO2 conversion and H2/CO ratio for dry reforming of methane over the 15% Ni/TiO2 (imp) catalyst and (c) CH4 conversion and CO selectivity for steam reforming reaction over the 15% Ni/TiO2 (sonic) catalyst. | ||
TGA analysis was used to estimate the deposited coke over the catalyst. Fig. 10 shows the TGA/DTA analysis performed in the presence of pure O2 of the spent catalyst on stream. The thermogram was divided into three different temperature regions. The first region below 260 °C showed a minimal weight loss and this can be ascribed to the loss of absorbed moisture and volatile species, such as reactants and products.42 The second region between 300 °C and 600 °C showed an increase in weight of the catalyst due to oxidation of the Ni particles. Finally, the third region above 600 °C showed a decrease in the weight of the catalyst due to oxidation of deposited coke with different degrees of graphitization. The amount of coke deposited on the spent catalyst was found to be around 0.6 wt%. The catalyst synthesized by sonochemical method showed less coke deposition due to stronger metal support interactions, contributing to the good reaction stability. Meanwhile, it has been reported that relatively large Ni particles are vulnerable to coke formation.43 Therefore, nanosized Ni particles and strong metal support interactions facilitate carbon gasification via H2O dissociation and oxygen spillover to Ni particles.
 | ||
| Fig. 10 TGA-DTA plot for the catalyst under pure oxygen after being on stream in the steam reforming reaction over 15% Ni/TiO2 (sonic) catalyst. | ||
5. Conclusions
Highly dispersed supported Ni nanoparticles over TiO2 were successfully prepared via a sonochemical assisted method and the performance of the material was investigated for dry and steam reforming of methane. The catalyst synthesized by the present approach exhibited excellent catalytic activity and stability for both the reactions compared to the catalyst prepared by incipient wetness impregnation method. For dry reforming, 86% CH4 and 84% CO2 conversions were obtained at 700 °C. Nearly 92% CH4 with 77% CO selectivity was observed at 700 °C for the steam reforming reaction. The high activity and stability of the catalyst is attributed to the fine dispersion (due to small metallic Ni clusters) of Ni species. The present catalyst is remarkably active and stable even after a long period and no appreciable coke deposition was observed. Therefore, the present approach is simple and feasible for the synthesis of supported metal catalysts, which can be used to enhance the catalytic performance.Acknowledgements
Authors gratefully acknowledge the financial support from Gas authority of India Limited.References
- J. Xu, C. M. Yeung, J. Ni, F. Meunier, N. Acerbi, M. Fowles and S. C. Tsang, Appl. Catal., A, 2008, 345, 119–127 CrossRef CAS PubMed.
- B. Höhlein, S. Von Andrian, T. Grube and R. Menzer, J. Power Sources, 2000, 86, 243–249 CrossRef.
- I. H. Son, S. J. Lee, A. Soon, H.-S. Roh and H. Lee, Appl. Catal., B, 2013, 134, 103–109 CrossRef PubMed.
- H.-S. Roh, K.-W. Jun, W.-S. Dong, S.-E. Park and Y.-S. Baek, Catal. Lett., 2001, 74, 31–36 CrossRef CAS.
- J. H. Jeong, J. W. Lee, D. J. Seo, Y. Seo, W. L. Yoon, D. K. Lee and D. H. Kim, Appl. Catal., A, 2006, 302, 151–156 CrossRef CAS PubMed.
- A. Lemonidou, M. Goula and I. Vasalos, Catal. Today, 1998, 46, 175–183 CrossRef CAS.
- T. Horiuchi, K. Sakuma, T. Fukui, Y. Kubo, T. Osaki and T. Mori, Appl. Catal., A, 1996, 144, 111–120 CrossRef CAS.
- Z. Hou, O. Yokota, T. Tanaka and T. Yashima, Catal. Lett., 2003, 87, 37–42 CrossRef CAS.
- M. Halabi, M. De Croon, J. Van der Schaaf, P. Cobden and J. Schouten, Appl. Catal., A, 2010, 389, 68–79 CrossRef CAS PubMed.
- N. Laosiripojana and S. Assabumrungrat, Appl. Catal., A, 2005, 290, 200–211 CrossRef CAS PubMed.
- S. Kurungot and T. Yamaguchi, Catal. Lett., 2004, 92, 181–187 CrossRef CAS.
- N. Laosiripojana, D. Chadwick and S. Assabumrungrat, Chem. Eng. J., 2008, 138, 264–273 CrossRef CAS PubMed.
- C. M. Kalamaras, K. C. Petallidou and A. M. Efstathiou, Appl. Catal., B, 2013, 136–137, 225–238 CrossRef CAS PubMed.
- P. S. Lambrou and A. M. Efstathiou, J. Catal., 2006, 240, 182–193 CrossRef CAS PubMed.
- C. Costa, S. Christou, G. Georgiou and A. Efstathiou, J. Catal., 2003, 219, 259–272 CrossRef CAS.
- R. Duarte, M. Nachtegaal, J. Bueno and J. V. Bokhoven, J. Catal., 2012, 296, 86–98 CrossRef PubMed.
- M. C. Bradford and M. A. Vannice, Appl. Catal., A, 1996, 142, 97–122 CrossRef CAS.
- N. Perkas, H. Rotter, L. Vradman, M. V. Landau and A. Gedanken, Langmuir, 2006, 22, 7072–7077 CrossRef CAS PubMed.
- J. Wei and E. Iglesia, J. Catal., 2004, 225, 116–127 CrossRef CAS PubMed.
- K. O. Christensen, D. Chen, R. Lødeng and A. Holmen, Appl. Catal., A, 2006, 314, 9–22 CrossRef CAS PubMed.
- M. Goula, A. Lemonidou and A. Efstathiou, J. Catal., 1996, 161, 626–640 CrossRef CAS.
- D. Ligthart, R. Van Santen and E. Hensen, J. Catal., 2011, 280, 206–220 CrossRef CAS PubMed.
- R. A. Van Santen, Acc. Chem. Res., 2008, 42, 57–66 CrossRef PubMed.
- H. S. Bengaard, J. K. Nørskov, J. Sehested, B. Clausen, L. Nielsen, A. Molenbroek and J. Rostrup-Nielsen, J. Catal., 2002, 209, 365–384 CrossRef CAS.
- L. Yin, Y. Wang, G. Pang, Y. Koltypin and A. Gedanken, J. Colloid Interface Sci., 2002, 246, 78–84 CrossRef CAS PubMed.
- Y. Wang, L. Yin and A. Gedanken, Ultrason. Sonochem., 2002, 9, 285–290 CrossRef CAS.
- P. Girault, J. Grosseau-Poussard, J. Dinhut and L. Marechal, Nucl. Instrum. Methods Phys. Res., 2001, 174, 439–452 CrossRef CAS.
- D. Dissanayake, M. P. Rosynek, K. C. C. Kharas and J. H. Lunsford, J. Catal., 1991, 132, 117–127 CrossRef CAS.
- Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy, ed. D. Briggs, M. P. Seah, D. Briggs and M. P. Seah, John Wiley & Sons, Chichester, 1983 Search PubMed.
- I. H. Son, S. J. Lee, A. Soon, H.-S. Roh and H. Lee, Appl. Catal. B., 2013, 134–135, 103–109 CrossRef CAS PubMed.
- D. R. Penn, J. Electron Spectrosc. Relat. Phenom., 1976, 9, 29–40 CrossRef CAS.
- J. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- N. Laosiripojana and S. Assabumrungrat, J. Power Sources, 2007, 163, 943–951 CrossRef CAS PubMed.
- J. Wei and E. Iglesia, J. Catal., 2004, 224, 370–383 CrossRef CAS PubMed.
- A. Berman, R. Karn and M. Epstein, Appl. Catal., A, 2005, 282, 73–83 CrossRef CAS PubMed.
- C. Carrara, J. Munera, E. Lombardo and L. Cornaglia, Top. Catal., 2008, 51, 98–106 CrossRef CAS.
- J. Wei and E. Iglesia, J. Phys. Chem. B, 2004, 108, 7253–7262 CrossRef CAS.
- M. Temkin, Adv. Catal., 1979, 28, 173–291 CrossRef CAS.
- J. G. Jakobsen, T. L. Jørgensen, I. Chorkendorff and J. Sehested, Appl. Catal., A, 2010, 377, 158–166 CrossRef CAS PubMed.
- C. A. Guimarães and M. d. Moraes, Appl. Catal., A, 2004, 258, 73–81 CrossRef PubMed.
- Z. Hou and T. Yashima, Appl. Catal., A, 2004, 261, 205–209 CrossRef CAS PubMed.
- P. Djinović, I. G. Osojnik Črnivec, B. Erjavec and A. Pintar, Appl. Catal., B, 2012, 125, 259–270 CrossRef PubMed.
- Y.-X. Pan, C.-J. Liu and P. Shi, J. Power Sources, 2008, 176, 46–53 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |