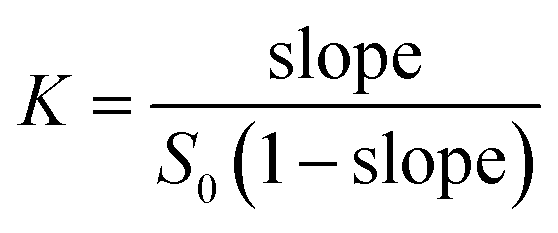DOI:
10.1039/C3RA45017A
(Paper)
RSC Adv., 2014,
4, 3348-3354
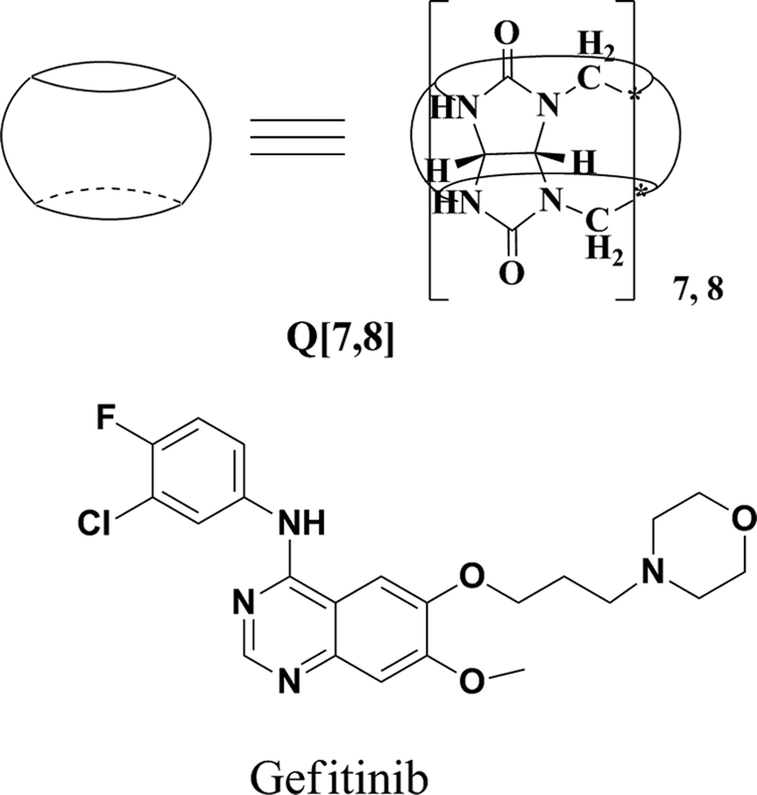
Cucurbit[7,8]urils binding to gefitinib and the effect of complex formation on the solubility and dissolution rate of the drug
Received
11th September 2013
, Accepted 25th November 2013
First published on 26th November 2013
Abstract
The host–guest interactions of macrocyclic hosts cucurbit[7,8]urils (Q[7,8]) with prototropic forms of the drug gefitinib (diprotonated: GH2+ and neutral: G) were investigated by electronic absorption spectroscopy. The two hosts Q[7,8] showed strong binding to the diprotonated form due to the ion–dipole interaction between the polar carbonyl oxygens of the hosts and the cationic GH2+. Q[7] and Q[8] showed similar binding to the neutral gefitinib, shifting its pKa by about 1.4 units and 0.7 units downward, respectively. Determination of the thermodynamic parameters of complex formation showed that the formation of inclusion complexes between gefitinib and Q[7,8] was enthalpy controlled, suggesting that hydrophobic and van der Waals interactions were the main driving forces. Moreover, the formation of inclusion complexes between Q[7] and Q[8] and gefitinib was confirmed by 1H NMR, which showed that the phenyl moiety of gefitinib was entrapped in the Q[7] cavity or likely located on the outside of the Q[7] cavity. The two models showed a fast dynamic equilibrium. Our results indicate that the complexation of gefitinib with Q[7] could enhance the solubility and dissolution rate of gefitinib in aqueous solution.
Introduction
Cucurbit[n]urils (Q[n]s) are highly symmetrical macrocyclic hosts comprised of glycoluril units linked through methylene bridges,and they possess highly polar carbonyl portals with a hydrophobic cavity.1–4 Q[n]s form remarkably stable complexes with a variety of guest molecules in aqueous solution through a combination of ion–dipole, hydrogen bonding, and hydrophobic interactions inside the cavity.5–8 Because the toxicity of Q[n]s is very low, they could be used safely as drug carriers.9,10 Recently, the possibility of using Q[n]s to delivery medicinal molecules has gained increasing attention. Q[n]s can be used as carriers to increase drug stability, control drug release, and increase the pharmaceutical and biological activity of certain compounds.11–14 Furthermore, the potential of cucurbit[n]uril molecules to modify the effective pKa values of included guests has generated increasing interest in recent studies.15–22 The complexation of drugs with Q[7] has been shown to shift their pKa, affecting their activation and stabilization while retaining their relevant chemical reactivity.23
Gefitinib(Fig. 1) is an epidermal growth factor receptor (EGFR) inhibitor that acts by interrupting EGFR signaling in target cells. It is used in the treatment of locally advanced or metastatic non-small cell lung cancer (NSCLC) in patients who have previously received chemotherapy. The molecule has pKa values of 5.4 and 7.2 and therefore ionizes progressively in solution as the pH falls. Gefitinib is soluble in aqueous solvents at pH 1, but is practically insoluble above pH 7, and its solubility drops sharply between pH 4 and pH 6. Such physicochemical properties may result in poor and variable oral absorption. Various approaches have been used to improve drug absorption in cases of limited solubility and dissolution rate: (1) micronization of drug particles, (2) nanoparticle formation, and (3) complexation of macrocyclic compounds with guest molecules.
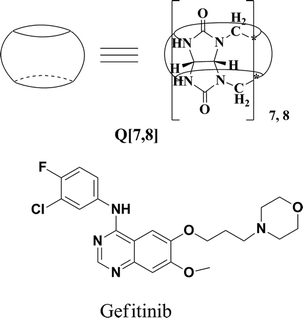 |
| | Fig. 1 Structures of Q[7,8] and gefitinib. | |
The use of host–guest complexes for improving the stability of drugs and facilitating their delivery and release has generated considerable interest. In the present study, we investigated the complexation of cucurbit[7,8]uril with protonated and neutral forms of gefitinib in aqueous solution and determined the pKa values of the included protonated and neutral forms to understand the effect of host–guest interactions on the prototropic equilibrium of the drug and its solubility and dissolution rate in aqueous solution.
Results and discussion
Absorption properties
In aqueous solution, gefitinib has two dissociation constants (pK1 = 5.4, pK2 = 7.2) and can exist in three different prototropic forms (diprotonated: GH2+, protonated: GH+, and neutral: G). Thus, in slightly acidic solution (pH ≈ 3), the diprotonated form (GH2+) of gefitinib prevails, having a characteristic absorption peak at 340 nm. On the other hand, the neutral form (G) of the drug dominates in alkaline solutions, showing its absorption peak at 331 nm.
Based on the ground state pKa of the drug, we adjusted the pH of the solution to approximately 3.0 to study the complexation of the GH2+ form (>99% abundance). Addition of Q[7] to the drug solution at pH 3.0 initially produced a broad absorption peak for the GH2+ form with a well-defined isosbestic point at 349 nm, and the peak absorbance subsequently decreased marginally up to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest ratio before attaining saturation at a Q[7] concentration of approximately 40 μM (Fig. 2a). This stoichiometry, which was confirmed by a Job's plot (insert of Fig. 2a). Notably, these spectral changes were distinctly different from the changes observed on GH2+ interaction with Q[8]. Addition of Q[8] solution resulted in significant changes: the absorption spectrum of GH2+ gradually shifted bathochromically by ∼10 nm, with a well-defined isosbestic point at ∼345 nm up to a 1:1 host–guest ratio (Fig. 2b and insert of Fig. 2b). These results suggested different binding mechanisms for these two homologue macrocycles. The binding constants were (1.24 ± 0.23) × 105 L mol−1 for the Q[7]–GH2+ system and (1.16 ± 0.14) × 106 L mol−1 for the Q[8]–GH2+ system.
:1 host–guest ratio before attaining saturation at a Q[7] concentration of approximately 40 μM (Fig. 2a). This stoichiometry, which was confirmed by a Job's plot (insert of Fig. 2a). Notably, these spectral changes were distinctly different from the changes observed on GH2+ interaction with Q[8]. Addition of Q[8] solution resulted in significant changes: the absorption spectrum of GH2+ gradually shifted bathochromically by ∼10 nm, with a well-defined isosbestic point at ∼345 nm up to a 1:1 host–guest ratio (Fig. 2b and insert of Fig. 2b). These results suggested different binding mechanisms for these two homologue macrocycles. The binding constants were (1.24 ± 0.23) × 105 L mol−1 for the Q[7]–GH2+ system and (1.16 ± 0.14) × 106 L mol−1 for the Q[8]–GH2+ system.
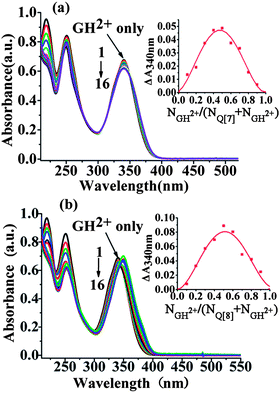 |
| | Fig. 2 Absorption spectra of the GH2+ form of gefitinib (40 μM) at different Q[7] (a) and Q[8] (b) concentrations and the corresponding ΔA ∼ NGH2+/(NGH2+ + NQ[7,8]) curve (inset) (pH = 3.0); [Q[7,8]]/μM: 1–16: 0; 8; 16; 24; 32; 40; 48; 56; 64; 72; 80; 88; 96; 106; 112; 120. | |
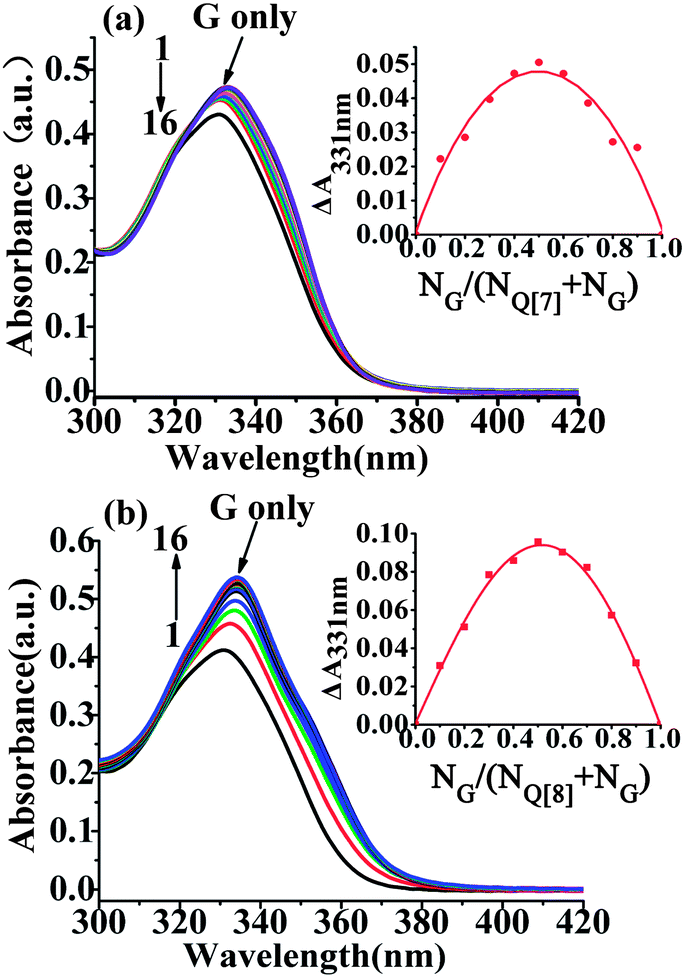
The interaction of the G form of the drug with Q[7] was analyzed at pH 9.7 (1 M NaOH). Upon addition of Q[7], the absorption peak of the G form of the drug underwent a bathochromic (from 331 nm to 333 nm) and small hyperchromic shift (Fig. 3a). The higher Q[7] concentrations required to obtain sizable changes in the absorption spectra indicated that the binding of the G form (at pH 9.7) was much lower than that of the GH2+ form (at pH 3.0). Similarly, the absorption peak of the G form underwent a red shift upon addition of Q[8] at pH 9.7 from 331 nm to 334 nm, along with a small increase in the peak absorbance (Fig.3b). These results indicate that the neutral G species forms a host–guest complex with Q[8]. The binding constants were (1.20 ± 0.02) × 105 L mol−1 for the Q[7]–G system, and (9.09 ± 0.54) × 105 L mol−1 for the Q[8]–G system.
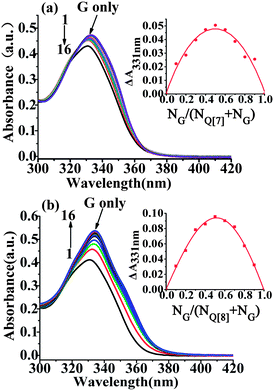 |
| | Fig. 3 Absorption spectra of the G form of gefitinib (40 μM) at pH 9.7 at different Q[7] (a) and Q[8] (b) concentrations and the corresponding ΔA ∼ NG/(NG + NQ[7,8]) curve (inset); [Q[7,8]]/μM: 1–16:0; 8;16; 24; 32; 40; 48; 56; 64; 72; 80; 88; 96; 106; 112; 120. | |
Thermodynamic parameters of the Q[7,8] inclusion complexes
To examine the interaction force between gefitinib and Q[7,8], the binding constants of the Q[7,8] inclusion complexes for the diprototropic forms of gefitinib (GH2+) were determined using spectrofluorimetric titrations at different temperatures, ranging from 298 K to 338 K with 10 K intervals (see Table 1 and Table 2). The plot of lnK as a function of T−1 is shown in Fig. 4. In the Q[7]@GH2+ system, the binding constant decreased gradually with temperature. The thermodynamic parameters for the complex were estimated using the van't Hoff equation (lnK = −ΔH/RT + ΔS/R) (Table 1). The changes in Gibbs free energy and enthalpy were negative, and entropy was positive. These data suggest that complex formation (ΔG = −29.06 ± 0.20 ∼ −32.83 ± 0.04 kJ mol−1) was largely driven by enthalpy (ΔH = −8.23 ± 0.30 kJ mol−1) in the presence of a favorable entropy (ΔS = 0.069 ± 0.00091 kJ mol−1 k−1). Therefore, complex formation was attributed to van der Waals and hydrophobic interactions. The Q[8]@GH2+ system showed a similar interaction force to the Q[7]@GH2+ system (see Table 2).
Table 1 Binding constants and thermodynamic parameters for the Q[7]@GH2+ complex at different temperatures
| T (K) |
K (L mol−1) |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (kJ mol−1 k−1) |
| 298 |
(1.24 ± 0.23) × 105 |
−29.06 ± 0.20 |
−8.23 ± 0.30 |
0.069 ± 0.00091 |
| 308 |
(1.09 ± 0.16) × 105 |
−29.70 ± 0.16 |
|
|
| 318 |
(9.84 ± 0.15) × 104 |
−30.40 ± 0.02 |
|
|
| 328 |
(9.10 ± 0.17) × 104 |
−31.14 ± 0.02 |
|
|
| 338 |
(8.29 ± 0.27) × 104 |
−32.83 ± 0.04 |
|
|
Table 2 Binding constants and thermodynamic parameters for the Q[8]@GH2+ complex at different temperatures
| T (K) |
K (L mol−1) |
ΔG (kJ mol−1) |
ΔH (kJ mol−1) |
ΔS (kJ mol−1 k−1) |
| 298 |
(1.16 ± 0.14) × 106 |
−34.60 ± 0.01 |
−14.13 ± 4.82 |
0.03 ± 0.00017 |
| 308 |
(7.14 ± 0.11) × 105 |
−34.51 ± 0.02 |
|
|
| 318 |
(4.92 ± 0.18) × 105 |
−34.65 ± 0.04 |
|
|
| 328 |
(3.88 ± 0.17) × 105 |
−35.09 ± 0.05 |
|
|
| 338 |
(2.58 ± 0.22) × 105 |
−35.02 ± 0.10 |
|
|
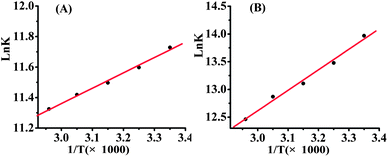 |
| | Fig. 4 Plot of lnK versus T−1 for the Q[7]@GH2+ complex (A) and the Q[8]@GH2+ complex (B). | |
Effect of Q[7,8] encapsulation on drug pKa values
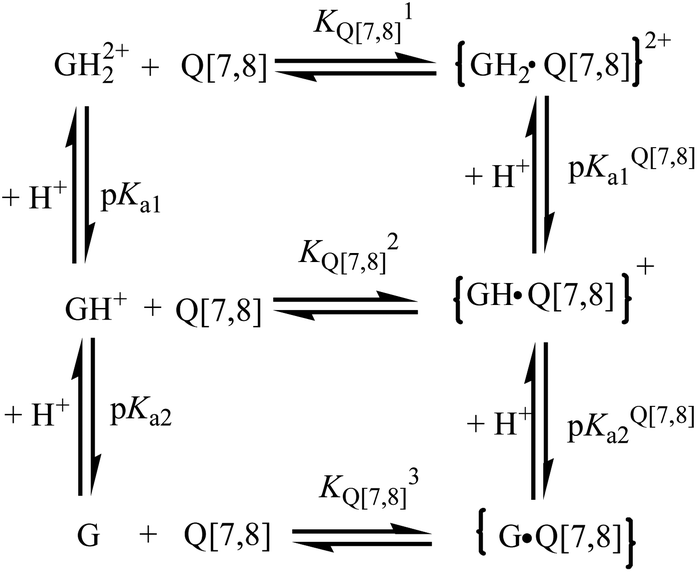
The UV-visible spectrum of gefitinib in this study was dependent on its protonation state in aqueous solution. Considering that both the GH2+ and G forms of gefitinib undergo sizable complexation with the Q[7,8] host, the prototropic equilibria for the Q[7,8]@gefitinib system can be represented by the thermodynamic model in Scheme 1, where pKa(1,2) and pKa(1,2)Q[7,8] represent the acid dissociation constants for the free and the complexed drug, respectively, and KQ[7,8],1 KQ[7,8]2 and KQ[7,8]3 represent the binding constants for the GH22+, GH+ and G forms of gefitinib with Q[7,8], respectively.
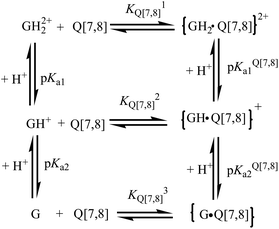 |
| | Scheme 1 Host–guest and acid dissociation equilibria in the complex formation between Q[7,8] and gefitinib (G) in aqueous solution. | |
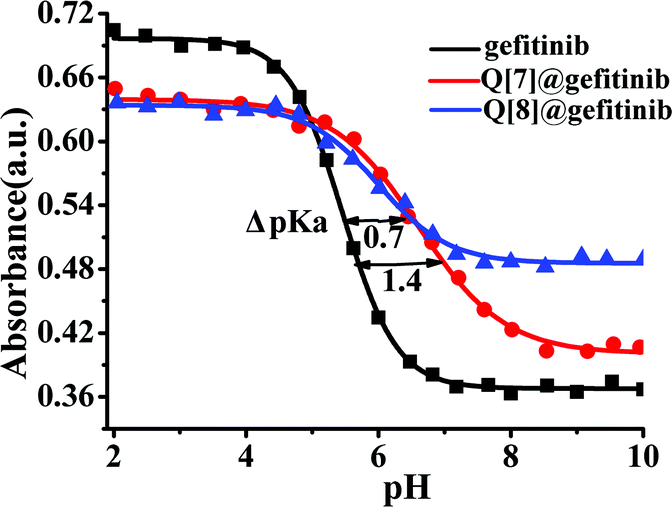
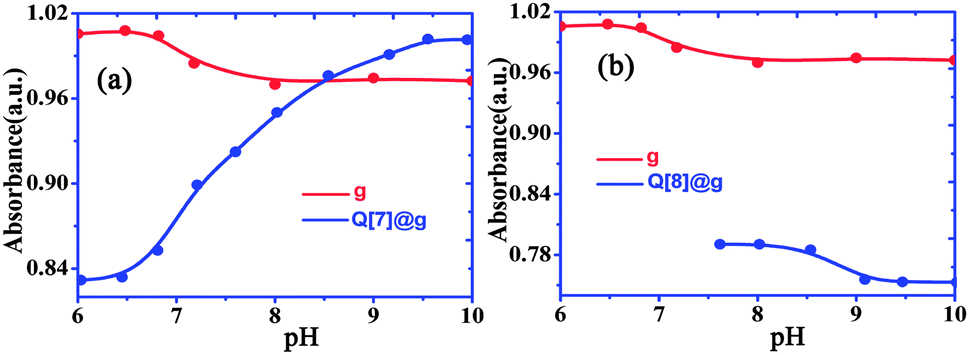
Free gefitinib exists as a monocationic species in neutral solutions with protonation at the quinazolin nitrogen group. In acidic solutions, the amine group undergoes protonation and the pKa1 value for the diprotonated gefitinib (GH22+) has been reported as 5.4, while the second acid dissociation to form the neutral guest occurs at a pKa2 value of 7.2.24 The changes in the absorption spectra of gefitinib at 340 nm as a function of the pH of the solution are shown in Fig. 5. The pKa of gefitinib in its ground state was determined to be 5.4 ± 0.1 from three independent measurements, well within the reported range of 5.4. The effect of the complex formation between GH22+ and Q[7,8] on the first acid dissociation constant was investigated by following the changes in the absorption spectra of the drug at 340 nm and 348 nm with a pH range of 2–10 in the presence of Q[7,8]. The results of the titration were pKa1Q[7] = 6.8 ± 0.1, ΔpKa = 1.4, pKa1Q[8] = 6.1 ± 0.1 and ΔpKa = 0.7. This pKa1Q[7,8] value is higher (upward shift by 0.6–1.3 units) than the pKa value of the free drug in aqueous solution. Thus, the complexation of gefitinib with Q[7,8] increases the basicity of the drug, as recently reported for other guests upon complexation by cucurbiturils and metal-organic supramolecular host structures.24 The titration of the {Q[7,8]@GH}+ complex with base in the pH range of 6–10 showed Fig. 6a and b. From the pH dependent UV spectral changes, the value of pKa2Q[7] and pKa2Q[8] was estimated to be 7.5 ± 0.2 and 8.8 ± 0.15.
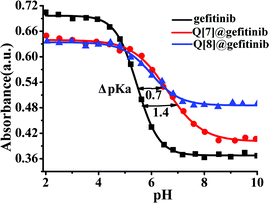 |
| | Fig. 5 UV pH titrations of gefitinib (■) (340 nm), Q[7]@gefitinib complexes (●) (340 nm) and Q[8]@gefitinib complexes (▲) (348 nm). | |
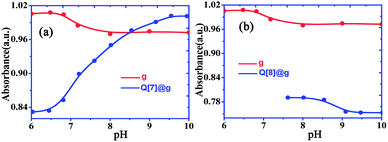 |
| | Fig. 6 Absorbance changes of Q[7]@GH complexes (a) and Q[8]@GH complexes (b) at different pH at λ = 248 nm. | |
1H NMR measurements
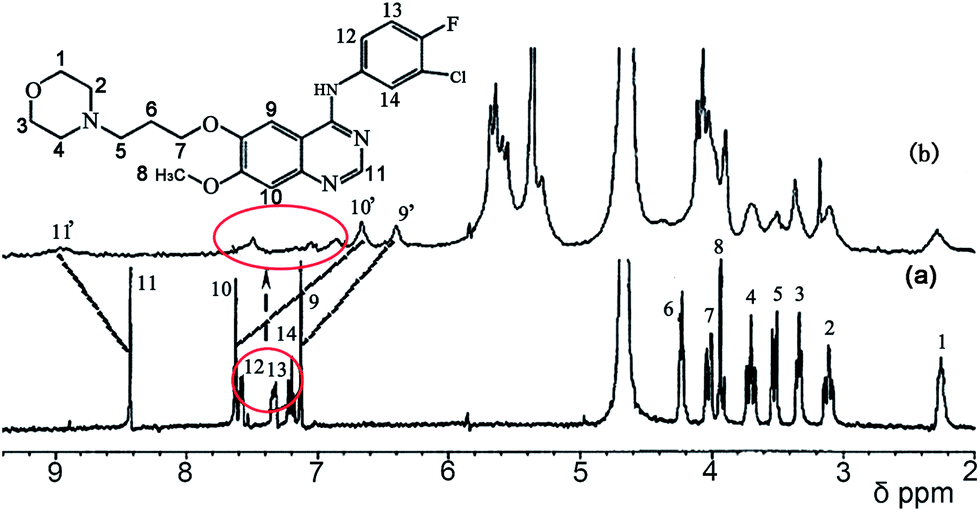
Fig. 7 shows the 1H NMR spectra of gefitinib in the absence (Fig. 7a) and in the presence (Fig. 7b) of a 1.0 equivalent of Q[7]. The undeuterated H1–H14 protons of gefitinib were detected. The H9 and H10 protons on the quinazolin moiety of gefitinib showed an upfield shift by ∼0.5 ppm and ∼0.3 ppm, respectively, and H11 on the quinazolin moiety exhibited a downfield shift of ∼0.1 ppm with 1.0 equivalent of Q[7], suggesting that Q[7] encapsulated part of the quinazolin moiety into its cavity. Moreover, the H1–4 protons on the morpholin ring and H5–8 protons of the alkyl chain also showed upfield shifts. These data indicated that the morpholin ring and alkyl chain moiety of gefitinib were also entrapped in the cavity of the host. Furthermore, these results showed that Q[7] encapsulated the quinazolin ring or the morpholin moiety of gefitinib into its cavity with a fast dynamic equilibrium. A possible inclusion model is depicted in Fig. 9 as Model-1. Furthermore, H11–13 of the phenyl moiety showed not only an upfield shift but also a downfield shift, suggesting that the phenyl moiety of gefitinib was entrapped in the cavity of Q[7] (Model-2) or likely located at the outside of the Q[7] cavity (Model-1). The two models reflect a fast dynamic equilibrium.
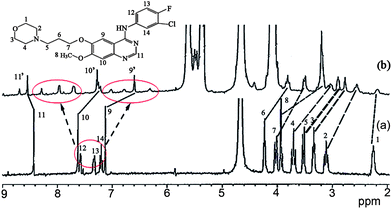 |
| | Fig. 7 1H NMR spectra (400 MHz, D2O) of gefitinib in the absence (a) and in the presence (b) of ∼1.0 equivalent of Q[7]. | |
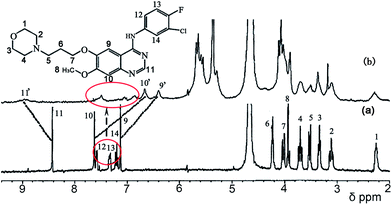 |
| | Fig. 8 1H NMR spectra (400 MHz, D2O) of gefitinib in the absence (a) and in the presence (b) of ∼0.4 equivalent of Q[8]. | |
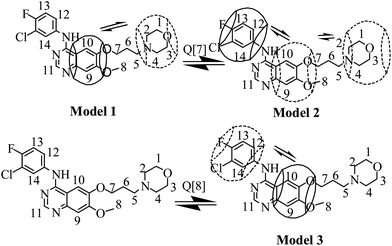 |
| | Fig. 9 The possible interaction modes between Q[7,8] and gefitinib. | |
When complexed with host Q[8], the quinazolin ring and phenyl moiety of the guest gefitinib were also entrapped in the cavity of the host. 1H NMR spectra of gefitinib in the absence and in the presence of Q[8] are shown in Fig. 8. The possible inclusion model is depicted in Fig. 9 as Model-3.
Effect of Q[7] on the solubility and dissolution rate of gefitinib
Gefitinib is soluble in aqueous solvents at pH 1, but is practically insoluble above pH 7 and its solubility drops sharply between pH 4 and pH 6. Therefore, in this study, the interactions between Q[7,8] and gefitinib were examined in the pH range of 4–10 with the phase solubility method.
Gefitinib is sparingly soluble in aqueous solvents at pH 5 with a solubility C0 of 0.047 mM. Phase solubility studies were conducted according to the method of Higuchi and Connors. The solubility of gefitinib increased linearly as a function of Q[7] concentration. Compared to the solubility of gefitinib in aqueous solvents in the absence of Q[7], linear increases in the solubility of gefitinib of up to 12-fold were achieved with increasing Q[7] concentrations up to 1.86 mM. The concentration of gefitinib in the presence of 1.86 mM Q[7] at pH 7 reached 0.59 mM, which is a 100-fold increase over that in water. A 130-fold increase in solubility was achieved in aqueous solvents at pH 9 in a 0.58 mM Q[7] solution (Table 3). Q[7] showed the highest solubilization efficiency for gefitinib. The formation of a complex between Q[7] and gefitinib may in part account for the increase in the solubility of the drug.
Table 3 Increase in the solubility of gefitinib in the presence of Q[7]. Seq is the binding gefitinib solubility in host–guest phase solubility equilibrium, S0 is the free gefitinib solubility
| Q[7] (mM) |
pH = 5 |
pH = 7 |
pH = 9 |
| Seq (mM) |
Seq/S0 |
Seq (mM) |
Seq/S0 |
Seq (mM) |
Seq/S0 |
| 0 |
0.047 |
1.00 |
0.0058 |
1.00 |
0.0044 |
1.00 |
| 0.37 |
0.13 |
2.77 |
0.11 |
18.97 |
0.12 |
27.27 |
| 0.75 |
0.24 |
5.11 |
0.22 |
37.93 |
0.24 |
54.55 |
| 1.12 |
0.36 |
7.66 |
0.39 |
67.24 |
0.37 |
84.09 |
| 1.49 |
0.47 |
10.00 |
0.48 |
82.76 |
0.50 |
113.64 |
| 1.86 |
0.58 |
12.34 |
0.59 |
101.72 |
0.58 |
131.82 |
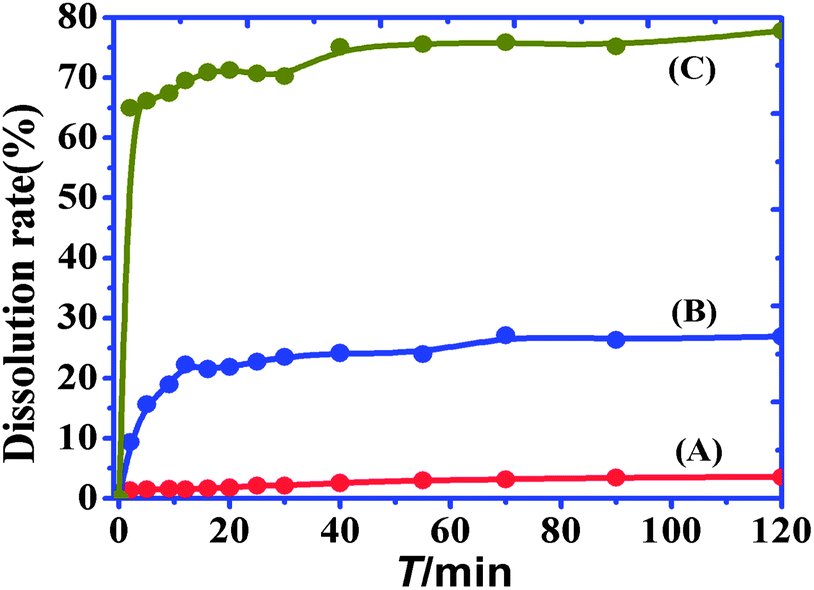
The dissolution profiles of the gefitinib and Q[7]@gefitinib systems are shown in Fig. 10. The physical mixture prepared with Q[7] did not have a significant effect on the dissolution rate of the drug, whereas the formation of inclusion complexes resulted in a significant improvement in the dissolution of the drug. The dissolution rates of the pure drug and physical mixtures at 20 min were 2% and 20%, respectively. The inclusion complex (1:1) showed rapid dissolution, with approximately 65% of the drug dissolved in 2 min.
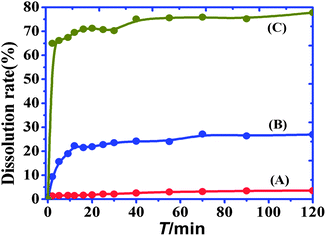 |
| | Fig. 10 The dissolution rate curves of gefitinib (A), Q[7]@gefitinib physical mixture (B) and inclusion complexes (C). | |
Cytotoxicity
To determine the effect of encapsulation on the anticancer activity of the host–guest complexes, gefitinib, Q[7,8]@gefitinib and Q[7,8] were tested using an in vitro growth inhibition assay in the A549 cell line (Table 4). Gefitinib encapsulation by Q[7] decreased cytotoxicity, and the cytotoxicity of Q[8]@gefitinib was similar to that of Q[7]@gefitinib. However, the reason for this result is not clear. Q[7,8] showed no cytotoxicity in the A549 cell line in in vivo tests (Table 4). We hope to elucidate the mechanism underlying the effect of cucurbiturils on drug cytotoxicity in the future.
Table 4 Cytotoxicity of gefitinib and Q[7,8]@gefitinib complexes(1:1) in the A549 cell line
| Sample |
IC50 (μM) |
| Adriamycin |
0.59 |
| Gefitinib |
6.79 |
| Q[7]@gefitinib complexes |
13.1 |
| Q[8]@gefitinib complexes |
12.9 |
| Q[7] |
>50 |
| Q[8] |
>50 |
Conclusions
The present study investigated the host–guest interactions of the drug gefitinib with the macrocyclic hosts Q[7,8]. The acid–base character of gefitinib was modified in a similar manner by Q[7,8], which shifted the pKa of the drug by approximately 1.4 and 0.7 units down, respectively. The thermodynamic parameters of the formation of inclusion complexes between gefitinib and Q[7,8] showed that it was enthalpy controlled, suggesting that hydrophobic and van der Waals interactions were the main driving forces. Moreover, the formation of inclusion complexes between Q[7] and Q[8] with gefitinib was confirmed by 1H-NMR, and the two models showed a fast dynamic equilibrium. Our results suggest that the complexation of gefitinib with Q[7] could be of value to improve the solubility and dissolution rate of gefitinib in aqueous solution.
Experimental section
Materials
Q[7] and Q[8] were prepared and purified according to previously published methods.3 Gefitinib was obtained from Sigma Aldrich and used as received. All other reagents were of analytical grade and were used as received. Double-distilled water was used for all experiments. The pH of the solution was adjusted with dilute hydrochloric acid or sodium hydroxide solutions.
UV-visible spectroscopy measurements
All UV-visible spectra were recorded in 1 cm quartz cells on an Agilent 8453 spectrophotometer, equipped with a thermostat bath (Hewlett Packard, California, USA). The pH was measured using a S-3C pH meter with an accuracy of ±0.05 (Dapu Instrument Co. Ltd, Shanghai, China). Stock solutions of gefitinib (1.00 × 10−3 mol L−1) were prepared by dissolving an appropriate amount of the drug in 0.1 M HCl and storing at 4 °C. Aqueous solutions of Q[7] (2.00 × 10−4 mol L−1) were prepared by dissolving the desired weights in distilled water. UV-visible spectra were obtained at 25 °C at a concentration of 4.00 × 10−5 mol L−1 gefitinib and different Q[7,8] concentrations in dilute hydrochloric acid (pH 2.0) for the Q[7,8]@GH2+ system and sodium hydroxide solutions (pH 9.7) for the Q[7,8]@G system. For each experiment, three repetitions were measured. Standard deviations were calculated on triplicate trials.
1H NMR measurements
To analyze the host–guest complexation of Q[7,8] and gefitinib, 2.0–2.5 × 10−3 mmol Q[7,8] in 0.5–0.7 mL D2O with Q[7,8]:gefitinib ratios ranging between 0 and 2 were prepared, and the corresponding 1H NMR spectra were recorded at 20 °C on a VARIAN INOVA-400 spectrometer.
pH solubility profiles
The same amounts of gefitinib, which were in large excess of the amount to reach saturation, were added to 50 mL of water. The pH was adjusted to various values at 25 °C with HCl and NaOH. The solutions were thermostatically shaken, allowed to settle, and filtered. The absorbance of the clear solution was then measured at 340 nm. The concentration of each solution was determined using appropriate calibration curves.
Phase solubility studies
Phase solubility was measured according to the method of Higuchi and Connors.25 Briefly, excess amounts of gefitinib were added to aqueous solutions containing various concentrations of Q[7] (0 to 2.0 mM). Samples were maintained at 25 °C, vibrated for 48 h, and incubated for 1 day until equilibrium was reached. Samples were then filtered (0.45 μm) and appropriately diluted with H2O. The absorbance at 340 nm was measured on an Agilent 8453 spectrophotometer. A gefitinib calibration curve was generated at pH 5. Apparent 1:1 stability constants (K) were determined from the initial part of the straight-line of the phase solubility diagrams:| |
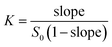 | (1) |
where (S0) is the free gefitinib aqueous solubility. The molar ratios of the complexes were determined by UV absorbance at 340 nm. Each experiment was conducted in triplicate.
Gibbs and Van't Hoff equations were used to estimate the thermodynamic parameters ΔH°, ΔS° and ΔG° according to the following formulas:
| | |
ΔG° = –RTlnK
| (2) |
| | |
lnK = ΔS°/R−ΔH°/RT
| (3) |
A plot of lnK versus T−1 produces: slope = −ΔH°/R and intercept = ΔS°/R.
Dissolution studies
Dissolution experiments were performed at 37 °C on a SRII 6-Flask Dissolution Test Station (Hanson Research, CA) according to Chinese Pharmacopoeia 2005 edition.26 Approximately 0.025 moles of gefitinib were added to 1 L of water in a 1 L beaker and paddle stirred at 50 rpm. At fixed time intervals (5, 10, 15, 20, 30, 60, 90, 120, 180, and 240 minutes), 10 mL samples were drawn with a syringe and spectrophotometrically assayed for gefitinib content as in the solubility studies. A correction was applied for the cumulative dilution caused by replacement of the sample with an equal volume of fresh medium. This procedure was performed again for the physical mixtures, inclusion complexes of gefitinib and cucurbit[7]uril. Each test was repeated three times, using samples containing an equivalent amount of gefitinib in each formulation.
Cytotoxicity assays
Cytotoxicity assays of gefitinib and Q[7,8]@gefitinib in the A549 cell line were performed using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) method.27 The A549 cell line was grown in RPMI 1640 medium supplemented with 10% fetal bovine serum and antibiotics and incubated at 37 °C with 5% CO2. Cells were plated in 96-well plates (100 μL per well) at a concentration of 5 × 104 cells per milliliter in complete medium and were then treated with a range of host–guest complex concentrations in 100 μL medium (0.00013–50 μM). The cells were incubated for 48 h under standard conditions before drug cytotoxicity was determined using an MTT-based growth inhibition assay. The assay was carried out by adding 20 μL MTT solution (5 mg mL−1) to the cell–drug mixtures and incubating for 4 h in standard conditions. The cells were harvested and mixed with 150 μL DMSO before the optical density (OD490) was read on a plate reader. The IC50 values were calculated using the SPSS software.
Acknowledgements
The work has been supported by the National Natural Science Foundation of China (Grant no. 21202026), the“Chun-Hui” Fund of Chinese Ministry of Education(Grant no. Z2011034), International cooperation projects of Science and Technology Agency of Guizhou Province(Grant no. 20127003) and Young Talent Development Project of Guizhou Province(Grant no. 2012154).
Notes and references
- W. A. Freeman, W. L. Mock and N. Y. Shih, J. Am. Chem. Soc., 1981, 103, 7367 CrossRef CAS.
- A. I. Day and A. P. Arnold, WO 0068232 2000, p. 8.
- J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540 CrossRef CAS.
- K. Kim, N. Selvapalam, Y. H. Ko, K. M. Park, D. Kim and J. Kim, Chem. Soc. Rev., 2007, 36, 267 RSC.
- A. I. Day, R. J. Blanch and A. P. Amold, Angew. Chem., Int. Ed, 2002, 41, 275 CrossRef CAS.
- R. J. Blanck, A. J. Sleeman, T. J. White, A. P. Arnold and A. I. Day, Nano Lett., 2002, 2, 147 CrossRef.
- J. X. Liu, Z. Tao, S. F. Xue, Q. J. Zhu and J. X. Zhang, Chin. J. Inorg. Chem., 2004, 20, 139 CAS.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844 CrossRef CAS PubMed.
- G. Hettiarachchi, D. Nguyen, J. Wu, D. Lucas, D. Ma, L. Isaacs and V. Briken, PloS ONE, 2010, 5, e10514 Search PubMed.
- V. D. Uzunova, C. Cullinane, K. Brix, W. M. Nau and A. I. Day, Org. Biomol. Chem., 2010, 8, 2037 CAS.
- N. J. Wheate, A. I. Day, R. J. Blanch, A. P. Arnold, C. Cullinane and J. G. Collins, Chem. Commun., 2004, 1424 RSC.
- M. S. Bali, D. P. Buck, A. J. Coe, A. I. Day and J. G. Collins, Dalton Trans., 2006, 5337 RSC.
- N. J. Wheate, D. P. Buck, A. I. Day and J. G. Collins, Dalton Trans., 2006, 451 RSC.
- Y. J. Zhao, M. S. Bali, C. Cullinane, A. I. Day and J. G. Collins, Dalton Trans., 2009, 5190 RSC.
- R. B. Wang, L. N. Yuan and D. H. Macartney, Chem. Commun., 2005, 5867 RSC.
- J. Mohanty, A. C. Bhasikuttan, W. M. Nau and H. Pal, J. Phys. Chem. B, 2006, 110, 5132 CrossRef CAS PubMed.
- R. B. Wang and D. H. Macartney, Org. Biomol. Chem., 2008, 6, 1955 CAS.
- M. Shaikh, J. Mohanty, P. K. Singh, W. M. Nau and H. Pal, Photochem. Photobiol. Sci., 2008, 7, 408 CAS.
- A. Praetorius, D. M. Bailey, T. Schwarzlose and W. M. Nau, Org. Lett., 2008, 10, 4089 CrossRef CAS PubMed.
- J. Wu and L. Isaacs, Chem.–Eur. J., 2009, 15, 11675 CrossRef CAS PubMed.
- I. W. Wyman and D. H. Macartney, Org. Biomol. Chem., 2010, 8, 247 CAS.
- M. Shaikh, S. D. Choudhury, J. Mohanty, A. C. Bhasikuttan and H. Pal, Phys. Chem. Chem. Phys., 2010, 12, 7050 RSC.
- N. Saleh, A. L. Koner and W. M. Nau, Angew. Chem. Int. Ed, 2008, 47, 5398 CrossRef CAS PubMed.
- M. H. Cohen, G. A. Williams, R. Sridhara, G. Chen, W. D. McGuinn, J. D. Morse, S. Abraham, A. Rahman, C. Y. Liang, R. Lostritto, A. Baird and R. Pazdur, Clin. Cancer Res., 2004, 10, 1212 CrossRef CAS.
- T. Higuchi and K. A. Connors, Adv. Anal. Chem. Instrum., 1965, 4, 117 CAS.
- Pharmacopoeia Commission, Chinese Pharmacopoeia 2005 Edition [M], Chemical Industry Press, Beijing, 2005, p. 801 Search PubMed.
- M. V. Berridge, P. M. Herst and A. S. Tan, Biotechnol. Annu. Rev., 2005, 11, 127 CAS.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.